P-ULVS:大規模バーチャルスクリーニング受託サービス
Preferred Networks は、Active Learning とドッキングシミュレーションを組み合わせることで、100 億規模の化合物ライブラリからの有望分子選抜を従来の約 1/1,500 の時間で可能にする受託サービス P-ULVS を提供している。
キーポイント
大規模ライブラリの創薬への貢献
100 億規模の超大規模化合物ライブラリを活用することで、新規骨格や高活性化合物の発見率が劇的に向上し、従来の手法では得られなかった成果が多数報告されている。
Active Learning による効率化
P-ULVS は全数探索(Brute force)やオンザフライ型ではなく、機械学習を用いて有望な化合物を順次選択・評価する Active Learning 方式を採用し、計算リソースを最適化する。
計算時間の劇的短縮
100 億規模の探索において、全数探索で約 317 年かかる計算を P-ULVS では約 2,000 時間(約 1/1,500)に短縮しつつ、有望化合物の約 80% を取得する実証結果がある。
コスト削減と迅速な合成
バーチャルスクリーニングにより実験コストを大幅に削減でき、Enamine REAL のように確立された反応に基づくライブラリを選抜することで、ヒット化合物の合成も迅速かつ安価に行える。
計算リソースの劇的短縮と高効率な探索
100億規模ライブラリの探索において、全数ドッキングに要する約317年を約2,000時間(約1/1,500)に短縮しつつ、有望化合物の約80%を取得できる性能を確認。
柔軟な化合物選択戦略のカスタマイズ
プロジェクト方針に応じて「ML Best」で最短サイクルでの取得や、「Uncertainty Sampling」による広範な探索、さらにLigand Efficiencyを用いた分子サイズ制御などを選択可能。
Baseline比較による再現率の推定手法
全数ドッキングが現実的でない大規模ライブラリに対し、独立したランダムサンプル(Baseline)との共通部分を利用することで、上位化合物の取得率(Recovery rate)を統計的に推定。
影響分析・編集コメントを表示
影響分析
本記事は、AI と計算科学を駆使した創薬プロセスの変革を示しており、従来の時間とコストの制約から解放された大規模化合物探索の実現可能性を具体的に証明しています。特に Active Learning の導入により、膨大な計算リソースを必要とする ULVS を実用的なスケールで運用可能にした点は、製薬業界全体の R&D パラダイムシフトに寄与する重要な技術的マイルストーンです。
編集コメント
PFN が提供する P-ULVS は、単なる計算速度の向上ではなく、AI の推論能力を活用して探索空間を賢く絞り込むことで、創薬のボトルネックである「ヒット発見」の効率化に決定的な役割を果たすサービスです。
本記事ではPreferred Networks (PFN)が提供する大規模バーチャルスクリーニング受託サービスP-ULVSの技術背景について紹介します。
はじめに
低分子創薬の初期段階であるヒット探索(Hit Identification)では、探索すべき化合物空間が極めて広大です。この広大な空間の中から、標的タンパク質に対して所望の活性を示す化合物を効率的に特定できるかどうかが、その後のヒット to リード、リード最適化に至る創薬プロセス全体の生産性を左右します。近年、試薬と化学反応の組み合わせから仮想的に列挙される、Enamine REALのような超大規模化合物ライブラリが整備されてきました。こうしたライブラリは収載化合物数が100億規模に達し、従来の1,000万規模のライブラリに比べてはるかに広い化合物空間をカバーします。このような膨大な数の化合物に対してタンパク質立体構造に対するドッキングシミュレーションを実行し、結合の可否(≒薬効)を判定する技術はUltra-Large Scale Virtual Screening(ULVS)と呼ばれています。PFNが提供するULVS受託サービスであるP-ULVSは、Active Learningとドッキングシミュレーションを組み合わせた技術であり、このような大規模化合物ライブラリから有望な化合物を効率的に取得します。
大規模ドッキングスクリーニングの背景と有効性
大規模バーチャルスクリーニングの有効性は、近年数多くの研究で報告されており、創薬のデファクトスタンダードになりつつあります。ライブラリサイズを100億規模まで拡大することには、明確なメリットと数々の実績が存在します。
まず挙げられるのが、新規骨格や高活性化合物の発見における顕著な成果です。たとえば、約1.38億化合物の探索によって、従来の300万化合物規模の探索では得られなかった高活性アゴニスト(< 1 nM)が取得された例や[1]、10億化合物以上の探索により標的タンパク質KEAP1に対して1 µM未満で結合する構造的に多様な分子群が同定された例が知られています[2]。
また、ライブラリの拡大はヒット率の大幅な改善にも直結します。実際に、ライブラリサイズを100万から1億規模に拡張することで、後続のHTS(High Throughput Screening)のヒット率が2〜5倍に改善した例が報告されています [2] 。さらに、100億規模の探索では、標準的手法(最大15%)を大きく上回る高いヒット率(CB2Rで33%、ROCK1で28.5%)を達成し、同時に新規骨格化合物が獲得された実績もあります[3]。こうした研究において、ドッキングスコアの最良値はライブラリサイズに対して対数スケールで改善し、その効果は10億規模に達してもなお飽和していないことが示されており、スケール拡大の重要性を裏付けています[4]。
さらに、コストと時間の削減という実用的な利点も見逃せません。バーチャルスクリーニングは実験的なHTSと比べてコストと時間を大幅に削減できるだけでなく、Enamine REALのように確立された合成反応に基づくライブラリから選抜すれば、ヒット化合物の合成も迅速かつ安価に行うことができます[3, 5]。
このように、より大規模なライブラリの活用は創薬成功率を劇的に向上させる可能性を秘めています。
P-ULVSの探索アプローチ
広大な化合物空間を探索するスクリーニング手法は、計算対象とする化合物の扱い方によって大きく3つに分類できます(表1)。
表1. 大規模ドッキングスクリーニングの分類
手法
データ形式
特徴
Brute force
enumerated
全件を探索するため網羅性が高いが、計算量が膨大
Building Block
on the fly
パーツ単位で評価し、スコアの加法性を前提とする。構造をon the flyで発生させるため全化合物データの保持は不要で、超広大なケミカルスペースの探索に適する
Active Learning
enumerated
機械学習でドッキング対象化合物を選択する。少ない計算量で高スコア領域を効率的に掘り当てる。全化合物データの保持が必要
まず1つ目は、列挙されたすべての化合物を全て計算する「全数探索(Brute force)」です。この手法は網羅性が高く取りこぼしがないという大きなメリットを持つ一方、計算量が膨大になるという課題があります。例えば、100億化合物すべてに対してドッキングを実施する場合、1化合物あたりわずか1秒で計算したとしても、単純計算で約317年もの時間を要してしまい、現実的ではありません。
2つ目は、パーツ(構成要素)単位で評価を行い、構造をその場で発生させる「Building Block(オン・ザ・フライ型)」です。この手法は全化合物データをあらかじめ保持しておく必要がないため、超広大な空間の探索に適しているという特徴があります。
そして3つ目が、機械学習モデルを用いて「結合が期待できる有望な化合物」をスマートに絞り込みながら探索を進める「Active Learning」です。ライブラリのごく一部を評価するだけで、高スコア領域に位置する有望化合物の大半を効率的に掘り当てることができます。
P-ULVSでは、これら3つのアプローチの中から、このActive Learning方式を採用しています。すでに列挙済みの大規模化合物ライブラリに対して、高い再現性で有望化合物を迅速に取得できるため、限られた計算リソースを最適化しつつ、100億規模という広大な化合物空間の恩恵を最大限に享受することが可能となります。
Active Learningを利用したドッキングスクリーニング
P-ULVSは、化合物の「選択」と「ドッキング」を反復的に繰り返すことで、大規模ライブラリの中から有望な化合物を効率的に選抜します。具体的には、以下の4つのステップを1サイクルとして、これをn回繰り返します(図1)。
選択: 機械学習モデルによる予測結果を基に、まだドッキングを実施していない化合物の中から次に評価すべき化合物を化合物ライブラリからx%選択します。初回サイクルでは予測モデルがないため、ランダムに選択します。
ドッキング: 選択された化合物に対してドッキングを実行し、ドッキングスコアを取得します。
トレーニング: ドッキングスコアが得られた化合物セットを用いて、機械学習モデルを作成します。
予測: 作成したモデルを用いて、化合物ライブラリ全件のドッキングスコアを予測します。
このサイクルを繰り返すことで、ドッキングスコアの良い領域へと探索が逐次的に誘導され、限られた計算リソースの中で有望な化合物群を効率的に取得できます。実際、ベンチマークの事例(5-HT2AR、100億規模ライブラリの探索)では、全数ドッキングにNVIDIA V100 1枚で約317年を要する計算を、P-ULVSでは約2,000時間(およそ1/1,500)に短縮しつつ(機械学習を含む)、結合が期待できる化合物の約80%を取得できることが確認されています。
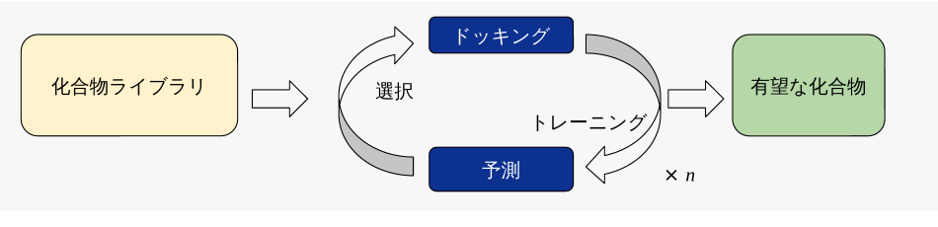
図1. P-ULVSにおけるActive Learning Dockingのワークフロー
化合物選択戦略
P-ULVSでは、Active Learningサイクルにおける化合物の選択方法(選択戦略)を、プロジェクトの方針に応じて柔軟にカスタマイズできます(表2)。標準的な「ML Best(Greedy)」戦略は、機械学習モデルがドッキングスコアの良いと予測した化合物を積極的に選択するもので、最短のサイクル数で有望化合物を取得することが期待できます。一方、「Uncertainty Sampling」戦略は予測精度が不十分な(確信度が低い)化合物を優先的に選択し、ケミカルスペースをより広く探索します。
さらに、Active Learningサイクルの中でドッキングスコアを補正し、選択する化合物の性質を制御することもできます。たとえば、化合物選択の基準にLigand Efficiency(LE、リガンド効率: 親和性を重原子数で規格化した指標)を採用することで、分子サイズが小さい化合物群からの活性化合物の取得が期待できます。
表2. 化合物選択戦略の実装例
選択方法
特徴
留意点
ML Best (Greedy)
ドッキングスコアが良いと予測された化合物を積極的に選択する。最短サイクルで活性化合物群を取得できる。
取得した骨格や化学的特徴が偏る可能性がある
Uncertainty Sampling
予測精度が不十分な化合物を選択し、ケミカルスペースを広く探索する。
探索のためのサイクルが必要
ベンチマーク取得方法
P-ULVSの性能を評価するため、セロトニン受容体5-HT2AR(PDB ID: 8UWL)を標的としたベンチマークテストを実施しました。化合物ライブラリにはEnamine REAL Database 2025.02の100億化合物を用い、Active Learningのサイクル数を4回(iter1, 2, 3, 4)、ドッキングスコアにはVina scoreを採用しました。1サイクル目はランダム100万化合物、2サイクル目以降はML Best (Greedy)戦略に切り替えて各100万化合物を選択、計400万化合物を評価しました(図2)。
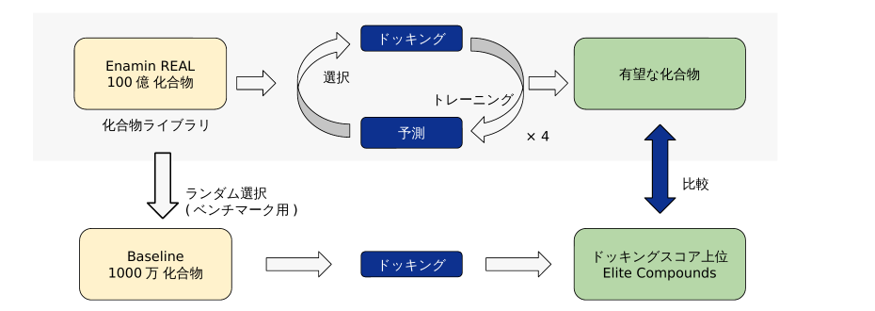
図2. ベンチマーク取得の流れ。P-ULVSで取得した有望化合物を、Baselineの上位化合物と比較する。
本検証では、シミュレーション上の妥当性を評価するため、「本来全数探索した際に見つかるはずの上位化合物を、P-ULVSがどれだけ漏らさず取得できたか(再現率:Recovery)」の推定値をパフォーマンスの指標としました。具体的なターゲットのイメージは図3の通りです。この例の場合、100億化合物のうち0.04%にあたる400万化合物をP-ULVSで選択・評価することで、ドッキングスコア上位0.001%のうち約80%を発見したというパフォーマンスを表します。
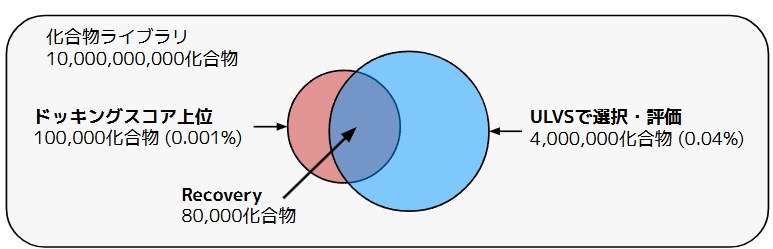
図3. Active Learning Dockingの概念図。ライブラリのごく一部の評価で、ドッキングスコア上位の大半を再現する。
ただし真のドッキングスコア上位を知るためには、全ての化合物のドッキングを実行し、事前に全スコアを知っておく必要があり、大規模ライブラリの場合にはこれは現実的ではありません。本検証ではあらかじめActive learningとは独立にランダムに選択したBaseline化合物セットを全数ドッキングし、パフォーマンスの推定に利用しています。統計学的に、このBaselineの化合物数が十分に大きければ、その中でのスコア上位集団は、オリジナルである100億化合物の母集団におけるスコア上位を代表したサンプルであると考えられます。本検証ではこのBaselineのスコア上位化合物と、P-ULVSの取得化合物を比較することで、P-ULVSの濃縮・探索性能を推定しています。
この考え方は図4のように、ドッキングスコア上位の化合物群をP、ULVSで選択・評価した化合物群をQとしたときに、Recovery rate \(n(P∩Q)/n(P)\)を、Baselineの集合Rとの共通部分を利用して、\(n(P∩Q∩R) / n(P∩R)\)と推定する方法になります。ここで\(P∩R\)は全数ドッキングしたRからスコア上位を選択することで推定します。
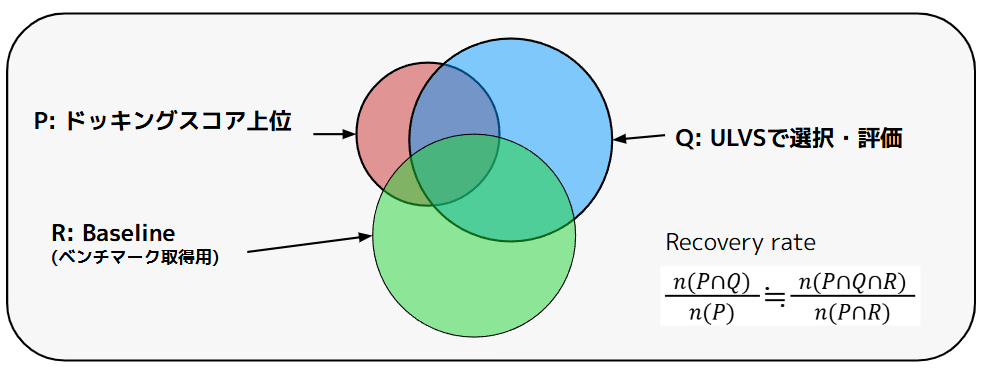
図4. ベンチマーク取得のためのBaseline。Baselineとの共通部分からRecovery rateを推定する。
ドッキングスコアの分布と上位化合物の取得
P-ULVSの各Active Learningサイクルで取得された化合物のドッキングスコア分布を図5に示します。初回サイクル(Iter 1)はランダム選択であるためスコアの分布は広く、結合が弱い化合物も多く含まれます。しかし2サイクル目以降、機械学習モデルによる選択(ML Best)が機能することで、取得される化合物のドッキングスコアが大幅に向上し(分布が左側=結合が強い側へシフト)、有望な化合物群へ濃縮されることが確認されました。
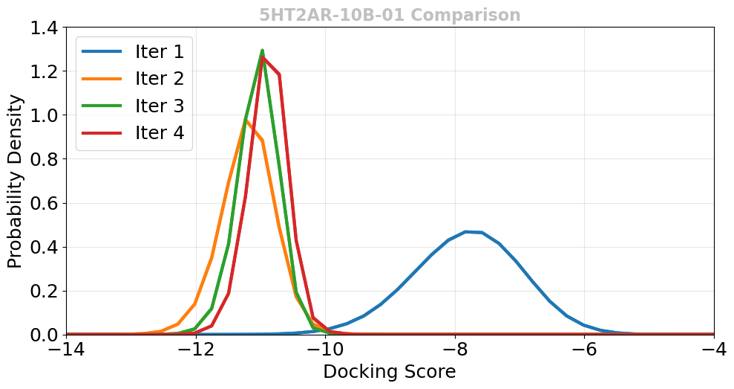
図5. P-ULVS各サイクルで取得した化合物のドッキングスコア分布
次に、別途全数ドッキングを実施したBaselineのドッキングスコア上位化合物を100ランクごとにグループ化し、P-ULVSが各グループの化合物をどの程度取得(再現)できたかを評価しました(図6)。その結果、ドッキングスコアが良い(上位の)化合物ほど高い取得率が得られることが示されました。すなわち、P-ULVSはライブラリのごく一部を評価するだけで、創薬研究で必要な上位の有望化合物を優先的に取得できることがわかります。
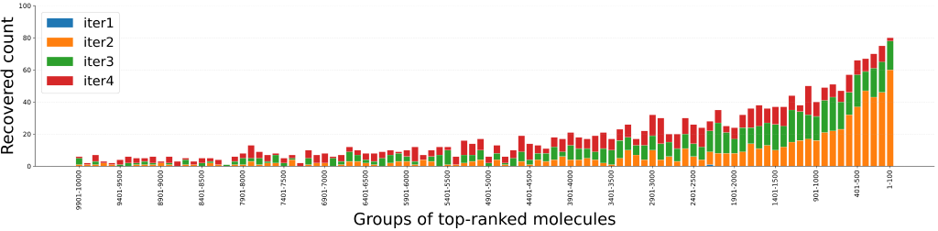
図6. Baselineのドッキングスコア上位化合物のULVSによる再現率
他のターゲットへの適用
P-ULVSは、立体構造が利用可能な様々な標的タンパク質に適用できます。前述の5-HT2ARに加え、CB2R(PDB ID: 5ZTY)、JAK3(5LWN)、ROCK1(2ETR)の各ターゲットについて、100億化合物のライブラリから400万化合物(0.04%)を評価した結果を表3に示します。いずれのターゲットでも、Baselineのドッキングスコア上位0.001%の化合物を約72〜81%という高い再現率で取得できており、P-ULVSが標的に依存せず安定した性能を発揮することが示されました。
表3. P-ULVSの計算事例(* Baselineの上位0.001%の再現率)
標的タンパク質(PDB ID)
ライブラリサイズ
P-ULVSで評価
再現率*
5-HT2AR (8UWL)
100億化合物
100万 ×4 (0.04%)
79.1%
CB2R (5ZTY)
100億化合物
100万×4 (0.04%)
75.7%
JAK3 (5LWN)
100億化合物
100万×4 (0.04%)
71.8%
ROCK1 (2ETR)
100億化合物
100万×4 (0.04%)
80.6%
まとめと展望
P-ULVSは、Active Learningによる効率的なケミカルスペース探索と分子ドッキングを組み合わせることで、従来は現実的ではなかった100億規模の大規模ライブラリのバーチャルスクリーニングを可能にします。本記事で紹介したベンチマークでは、ライブラリ全体のわずか0.04%を評価することで、ドッキングスコア上位の有望化合物の大半を効率的に取得できることを示しました。これは、創薬研究初期のヒット探索において、有望化合物を発見するチャンスを大きく広げ、実験コストや時間を削減し、高難易度ターゲットへの適用や新規骨格の発見をも可能にするものです。
今後は、選択戦略のさらなる高度化や、より広大な化合物空間への対応を進め、創薬の探索プロセス全体の生産性向上に貢献していきます。有望な化合物の更なる絞り込みやその後の合成展開デザインの評価には、より高精度な分子シミュレーションが必要です。結合自由エネルギー計算P-FEPについての記事もぜひご覧ください[6, 7]。
—
本記事およびP-ULVSに関するお問い合わせ先:drug-info[a]preferred.jp
[a]を @ に置き換えてご送信ください。
———
References
Lyu, J., et al. (2019). “Ultra-large library docking for discovering new chemotypes.” Nature 566 (7743), 224-229.
Gorgulla, C., et al. (2020). “An open-source drug discovery platform enables ultra-large virtual screens.” Nature 580(7805): 663-668.
Sadybekov, A. A., et al. (2022). “Synthon-based ligand discovery in virtual libraries of over 11 billion compounds.” Nature 601(7893): 452-459.
Lyu, J., et al. (2023). “Modeling the expansion of virtual screening libraries.” Nat Chem Biol 19 (6), 712-718.
Gorgulla, C. (2023). “VirtualFlow 2.0 – The Next Generation Drug Discovery Platform Enabling Adaptive Screens of 69 Billion Molecules.” bioRxiv. https://doi.org/10.1101/2023.04.25.537981
P-FEP (RBFEP計算サービス) 提供開始, Preferred Networks社ブログ https://tech.preferred.jp/ja/blog/pfep-launch/
Active Learning駆動型RBFEPによる実践的リード化合物最適化の新戦略, Preferred Networks社ブログ https://tech.preferred.jp/ja/blog/al-fep/
投稿 P-ULVS: 大規模バーチャルスクリーニング受託サービス は Preferred Networks Tech Blog に最初に表示されました。
関連記事
創薬DMTAサイクルの高速化! Active Learning駆動型Relative Binding FEP(RBFEP)による「実践的」リード化合物最適化の新戦略
Preferred Networksが、Free Energy Perturbation(FEP)に基づき、Active Learningを駆動したRelative Binding FEP(RBFEP)という新手法を開発した。この手法は、創薬プロセスにおけるDMTAサイクルを高速化し、実践的なリード化合物最適化を可能にする。
構造認識pLMを用いた化合物-タンパク質相互作用予測手法の開発
PFNリサーチャーの武本氏らが、タンパク質の立体構造と化合物の物性情報を統合した言語モデルを用いて、化合物とタンパク質の相互作用を予測する手法を開発した。
評価指標に依存する注釈飽和:ラベル分布からの学習における研究
Apple Machine Learning は、注釈者の不一致が信号となり得ることを示し、評価指標によって必要な注釈者数が異なる「注釈飽和」を特定した。NLI モデルの実験により、不一致を検出するエントロピー相関には約 20〜50 人の注釈者が必要であることが明らかになった。
今日のまとめ
AI日報で今日の重要ニュースをまとめ読み