潜在拡散を用いた生成のためのタンパク質折りたたみモデルの再利用
Berkeley AI Research は、タンパク質折りたたみモデルの潜在空間を活用し、配列と構造を同時に生成するマルチモーダル生成モデル「PLAID」を発表した。
キーポイント
マルチモーダル同時生成の実現
既存モデルが骨格のみを生成するのに対し、PLAID は離散的なアミノ酸配列と連続的な全原子構造座標を同時に生成できる。
配列データのみでの学習
構造データベースよりも桁違いに多い配列データベースのみを用いてトレーニング可能であり、学習データの制約を大幅に緩和した。
テキストによる制御インターフェース
機能や生物種(ヒト化など)といったコンポーザショナルなプロンプトを通じて、目的に応じた有用なタンパク質を生成する制御を実現している。
シーケンスデータのみでのトレーニング
構造データベースよりも桁違いに大きいシーケンスデータベースを活用し、実験的構造の取得コストを回避して生成モデルを訓練可能。
潜在空間拡散とデコーディング
タンパク質折りたたみモデル(ESMFold)の潜在空間で拡散モデルを学習し、サンプリング後に凍結された重みを用いて構造を復元する。
CHEAPによる潜在空間圧縮
Transformerベースモデルの潜在空間が巨大かつスパースである課題に対し、CHEAP手法でシーケンスと構造の結合埋め込みを圧縮し、高解像度生成を実現。
汎用的なマルチモーダル生成への拡張可能性
本手法はタンパク質のシーケンスと構造に限定されず、豊富なモダリティから希少なモダリティへの予測器が存在するあらゆる分野でマルチモーダル生成に応用可能です。
影響分析・編集コメントを表示
影響分析
この研究は、タンパク質生成AIが単なる予測モデルから、創薬や材料設計に直接活用可能な設計ツールへと進化することを示唆しています。特に、構造データに依存せず配列データのみで学習可能である点は、未知のタンパク質デザインにおけるデータ不足というボトルネックを解決する画期的なアプローチです。
編集コメント
ノーベル賞受賞直後のこの発表は、AI が生物学において「予測」から「創造」へとパラダイムシフトを起こしたことを象徴する出来事と言えます。特に学習データの制約を突破した点は、今後のバイオインフォマティクス分野における実用化への大きな一歩です。
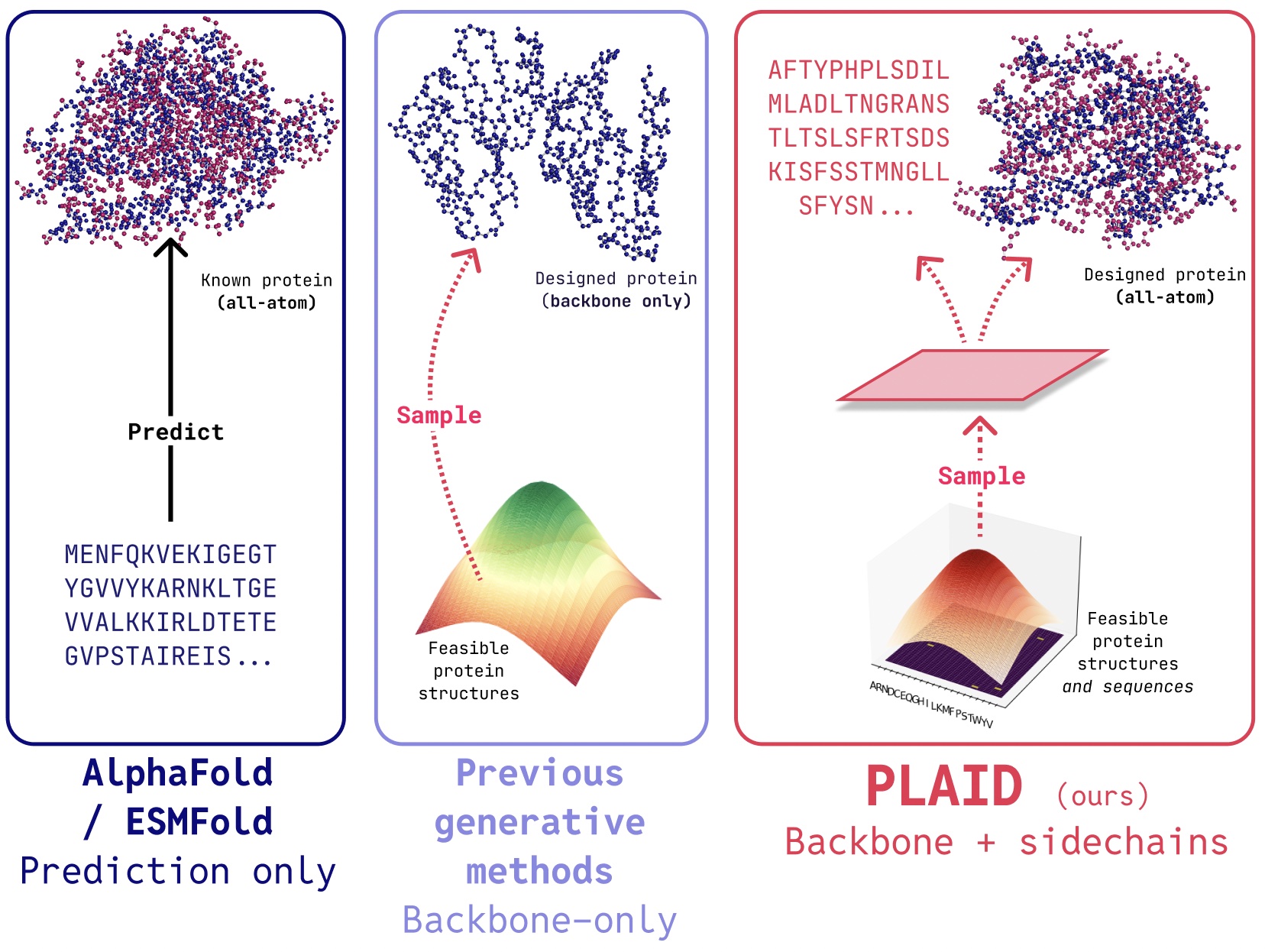 PLAIDは、タンパク質折り畳みモデルの潜在空間を学習することで、タンパク質の1次元配列と3次元構造を同時に生成するマルチモーダル生成モデルです。
PLAIDは、タンパク質折り畳みモデルの潜在空間を学習することで、タンパク質の1次元配列と3次元構造を同時に生成するマルチモーダル生成モデルです。
2024年のノーベル賞がAlphaFold2に授与されたことは、生物学におけるAIの役割に対する認識の重要な瞬間を示しています。タンパク質折り畳みの次に来るものは何でしょうか?
PLAIDでは、タンパク質折り畳みモデルの潜在空間からサンプリングすることを学習し、新しいタンパク質を生成する手法を開発しました。これは、構成可能な機能や生物種のプロンプトを受け入れられ、構造データベースよりも2〜4桁大きい配列データベースで学習することができます。多くの従来のタンパク質構造生成モデルとは異なり、PLAIDはマルチモーダル共生成の問題設定、すなわち離散的な配列と連続的な全原子構造座標の両方を同時に生成する問題に取り組みます。
構造予測から実世界の薬剤設計へ
拡散モデルがタンパク質を生成する能力について有望であることが最近の研究で示されていますが、実世界の応用を非現実的にするような従来モデルの限界が依然として存在します。例えば:
全原子生成:既存の生成モデルの多くは、主鎖原子のみを生成します。全原子構造を生成し、側鎖原子を配置するためには、配列を知る必要があります。これは、離散的モダリティと連続的モダリティの同時生成を必要とするマルチモーダル生成問題を生み出します。
生物種特異性:ヒトでの使用を目的としたタンパク質医薬品は、ヒト免疫系によって破壊されるのを避けるために、ヒト化される必要があります。
制御仕様:創薬とそれを患者の手に渡すことは複雑なプロセスです。これらの複雑な制約をどのように指定すればよいでしょうか?例えば、生物学的问题が解決された後でも、錠剤がバイアルよりも輸送が容易だと判断し、溶解性に関する新たな制約を追加するかもしれません。
「有用な」タンパク質の生成
単にタンパク質を生成するよりも、生成を制御して有用なタンパク質を得ることの方が重要です。そのためのインターフェースはどのようなものになるでしょうか?
 インスピレーションとして、構成可能なテキストプロンプトによる画像生成の制御方法を考えてみましょう(Liu et al., 2022の例)。
インスピレーションとして、構成可能なテキストプロンプトによる画像生成の制御方法を考えてみましょう(Liu et al., 2022の例)。
PLAIDでは、この制御仕様のインターフェースを模倣しています。最終目標はテキストインターフェースを介して完全に生成を制御することですが、ここでは概念実証として、機能と生物種という2つの軸に関する構成可能な制約を考察します:
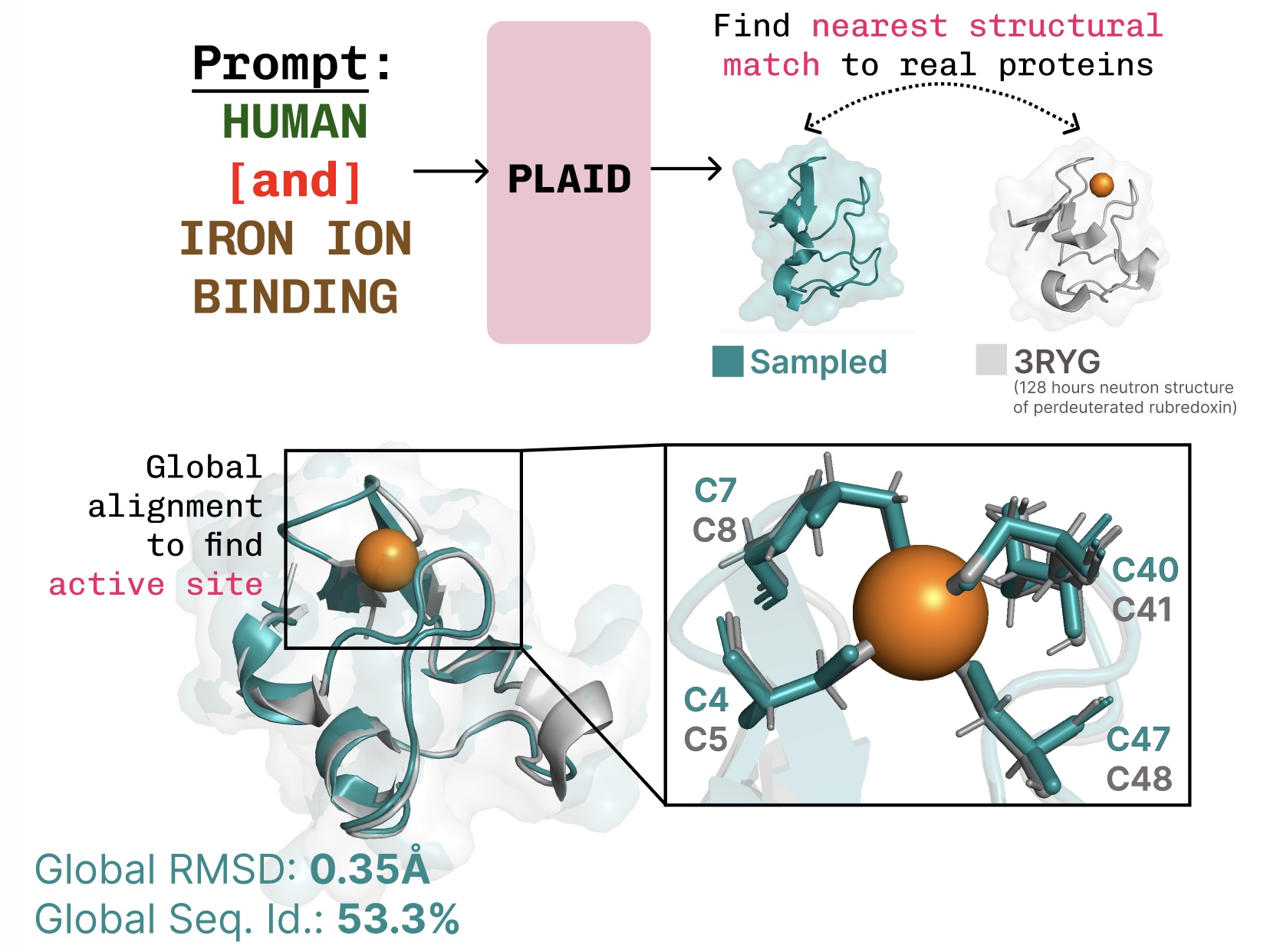 機能-構造-配列の関連性の学習。PLAIDは、金属タンパク質でよく見られる四面体システイン-Fe2+/Fe3+配位パターンを学習しながら、高い配列レベルの多様性を維持します。
機能-構造-配列の関連性の学習。PLAIDは、金属タンパク質でよく見られる四面体システイン-Fe2+/Fe3+配位パターンを学習しながら、高い配列レベルの多様性を維持します。
配列のみの学習データを用いた訓練
PLAIDモデルのもう一つの重要な側面は、生成モデルを訓練するために配列のみが必要であることです!生成モデルは、その学習データによって定義されるデータ分布を学習します。実験的な構造を得るよりも配列を得る方がはるかにコストが低いため、配列データベースは構造データベースよりもかなり大規模です。
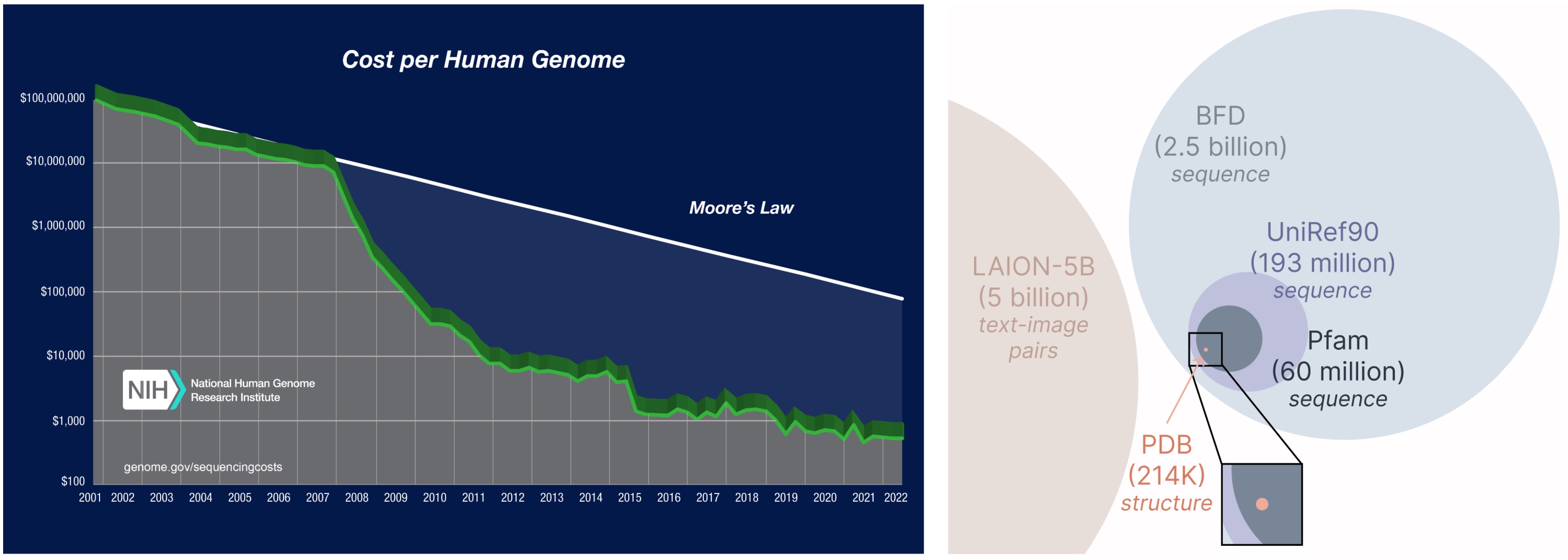 より大きく広範なデータベースからの学習。タンパク質配列を取得するコストは、実験的に構造を決定するコストよりもはるかに低く、配列データベースは構造データベースよりも2〜4桁大きいです。
より大きく広範なデータベースからの学習。タンパク質配列を取得するコストは、実験的に構造を決定するコストよりもはるかに低く、配列データベースは構造データベースよりも2〜4桁大きいです。
どのように機能するのか?
生成モデルを訓練して、配列データのみを使用して構造を生成できる理由は、タンパク質折り畳みモデルの潜在空間上で拡散モデルを学習するためです。その後、推論時に、有効なタンパク質のこの潜在空間からサンプリングした後、タンパク質折り畳みモデルの凍結された重みを使用して構造をデコードできます。ここでは、AlphaFold2モデルの後継であり、検索ステップをタンパク質言語モデルに置き換えたESMFoldを使用します。
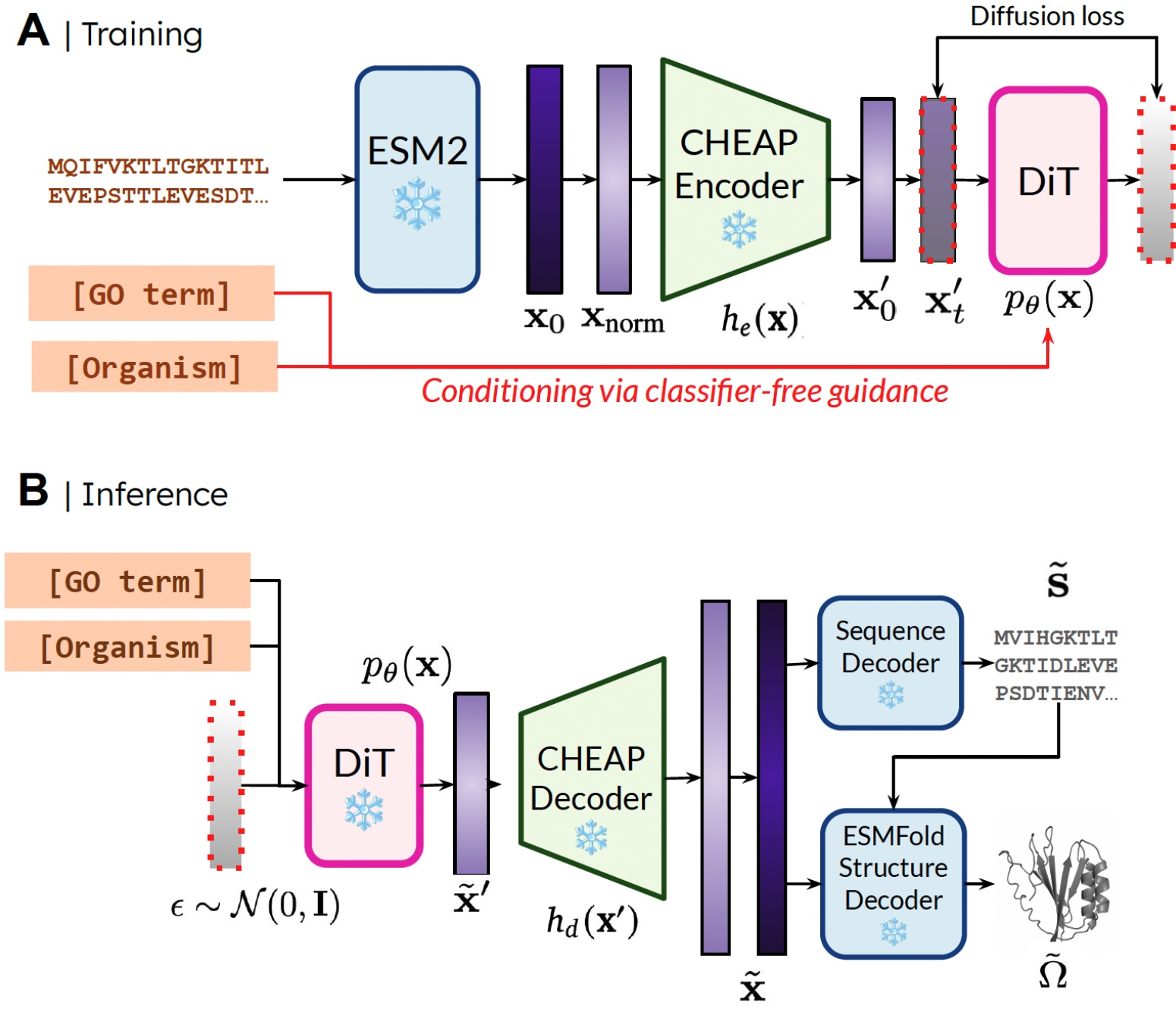 我々の手法。訓練時には、埋め込みを得るために配列のみが必要です。推論時には、サンプリングされた埋め込みから配列と構造をデコードできます。❄️は凍結された重みを示します。
我々の手法。訓練時には、埋め込みを得るために配列のみが必要です。推論時には、サンプリングされた埋め込みから配列と構造をデコードできます。❄️は凍結された重みを示します。
このようにして、事前学習されたタンパク質折り畳みモデルの重みに含まれる構造的理解情報を、タンパク質設計タスクに利用できます。これは、ロボティクスにおける視覚-言語-行動(VLA)モデルが、インターネット規模のデータで訓練された視覚-言語モデル(VLM)に含まれる事前情報を利用して知覚、推論、理解情報を提供する方法と類似しています。
タンパク質折り畳みモデルの潜在空間の圧縮
この方法を直接適用する際の小さな問題点は、ESMFoldの潜在空間、実際には多くのトランスフォーマーベースモデルの潜在空間が、多くの正則化を必要とすることです。この空間はまた非常に大きいため、この埋め込みを学習することは、高解像度画像合成へのマッピングになってしまいます。
この問題に対処するため、我々はまたCHEAP(Compressed Hourglass Embedding Adaptations of Proteins)を提案します。これは、タンパク質配列と構造の結合埋め込みのための圧縮モデルを学習するものです。
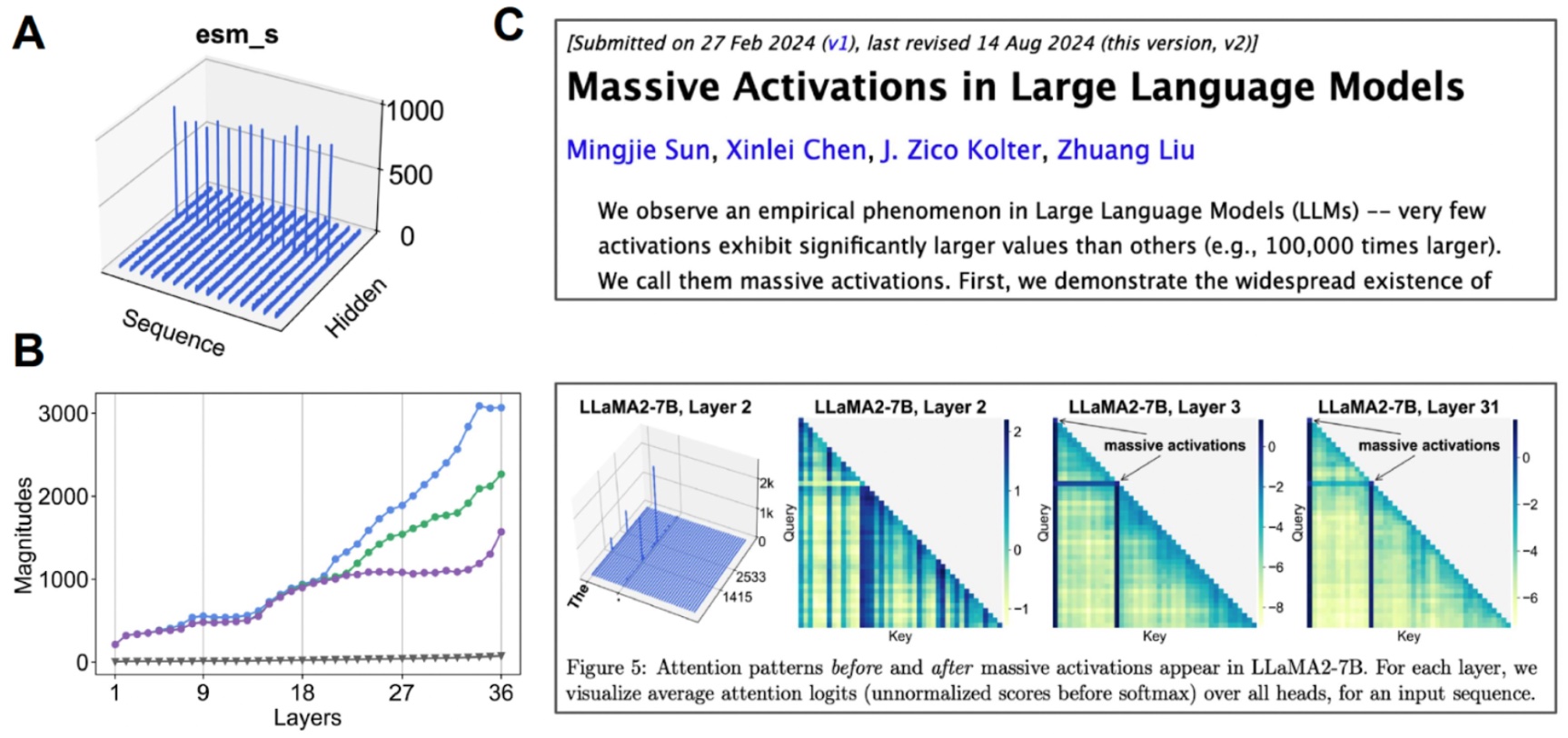 潜在空間の調査。(A)各チャネルの平均値を可視化すると、一部のチャネルが「大規模な活性化」を示します。(B)中央値(灰色)と比較して上位3つの活性化を調べ始めると、これが多くの層で起こっていることがわかります。(C)大規模な活性化は、他のトランスフォーマーベースモデルでも観察されています。
潜在空間の調査。(A)各チャネルの平均値を可視化すると、一部のチャネルが「大規模な活性化」を示します。(B)中央値(灰色)と比較して上位3つの活性化を調べ始めると、これが多くの層で起こっていることがわかります。(C)大規模な活性化は、他のトランスフォーマーベースモデルでも観察されています。
この潜在空間は実際には高度に圧縮可能であることがわかりました。我々が扱っている基本モデルをよりよく理解するために、少しメカニズム的解釈可能性を行うことで、全原子タンパク質生成モデルを作成することができました。
この研究ではタンパク質配列と構造の生成の場合を検討していますが、この方法は、より豊富なモダリティからより乏しいモダリティへの予測器が存在する任意のモダリティに対して、マルチモーダル生成を実行するように適応させることができます。タンパク質の配列から構造への予測器が、より複雑なシステム(例えば、AlphaFold3は核酸や分子リガンドと複合体を形成したタンパク質も予測可能)に取り組み始めているため、同じ方法を使用してより複雑なシステム上でマルチモーダル生成を実行することは容易に想像できます。我々の方法を拡張するための共同研究、またはウェットラボで我々の方法をテストすることに興味がある方は、ぜひご連絡ください!
我々の論文があなたの研究に役立った場合は、PLAIDとCHEAPについて以下のBibTeXの使用をご検討ください:
@article{lu2024generating, title={Generating All-Atom Protein Structure from Sequence-Only Training Data}, author={Lu, Amy X and Yan, Wilson and Robinson, Sarah A and Yang, Kevin K and Gligorijevic, Vladimir and Cho, Kyunghyun and Bonneau, Richard and Abbeel, Pieter and Frey, Nathan}, journal={bioRxiv}, pages={2024--12}, year={2024}, publisher={Cold Spring Harbor Laboratory} }
@article{lu2024tokenized, title={Tokenized and Continuous Embedding Compressions of Protein Sequence and Structure}, author={Lu, Amy X and Yan, Wilson and Yang, Kevin K and Gligorijevic, Vladimir and Cho, Kyunghyun and Abbeel, Pieter and Bonneau, Richard and Frey, Nathan}, journal={bioRxiv}, pages={2024--08}, year={2024}, publisher={Cold Spring Harbor Laboratory} }
プレプリント(PLAID, CHEAP)とコードベース(PLAID, CHEAP)もご覧いただけます。
ボーナス:タンパク質生成の楽しみ!
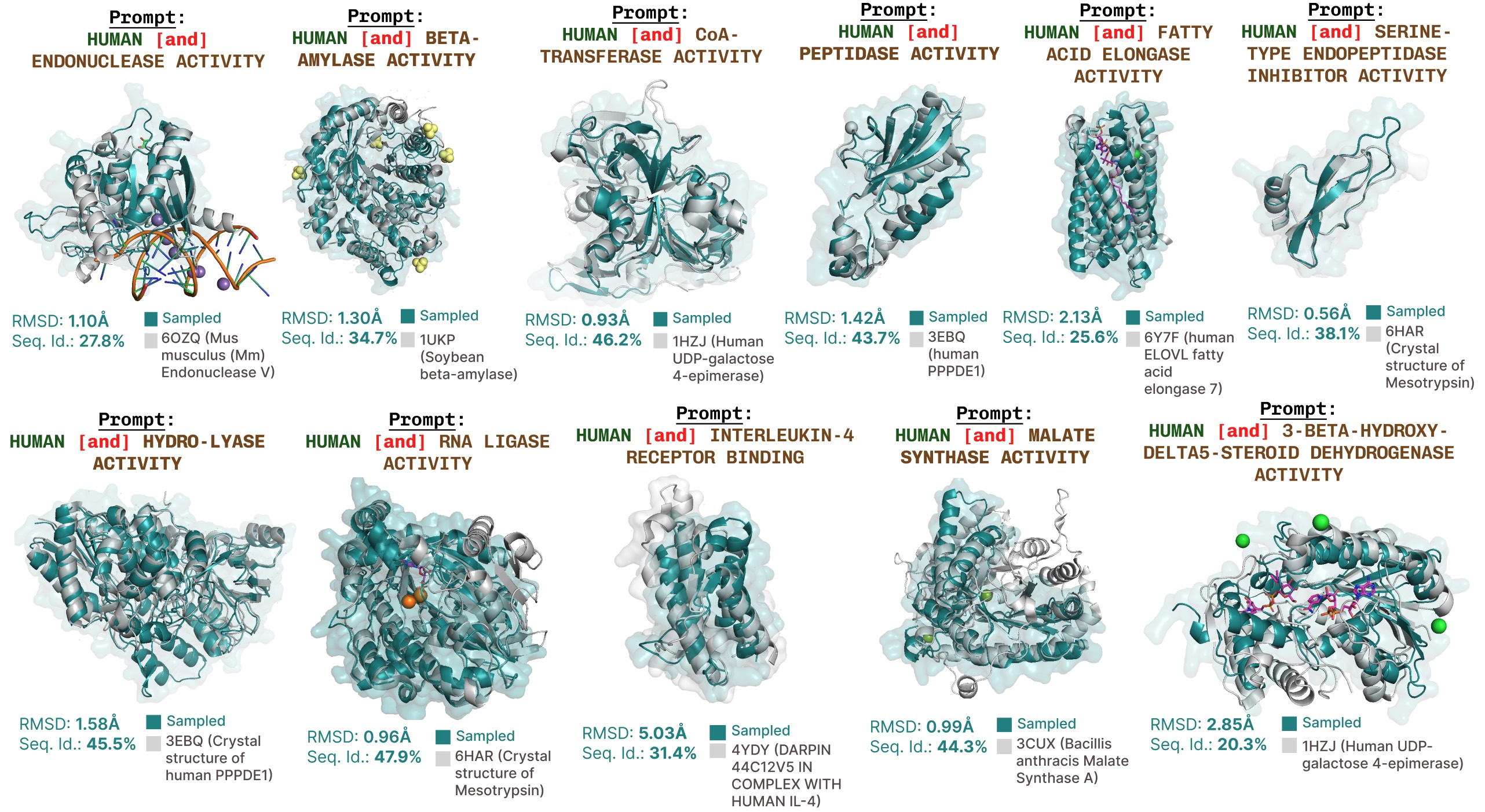 PLAIDによる追加の機能プロンプト生成。
PLAIDによる追加の機能プロンプト生成。
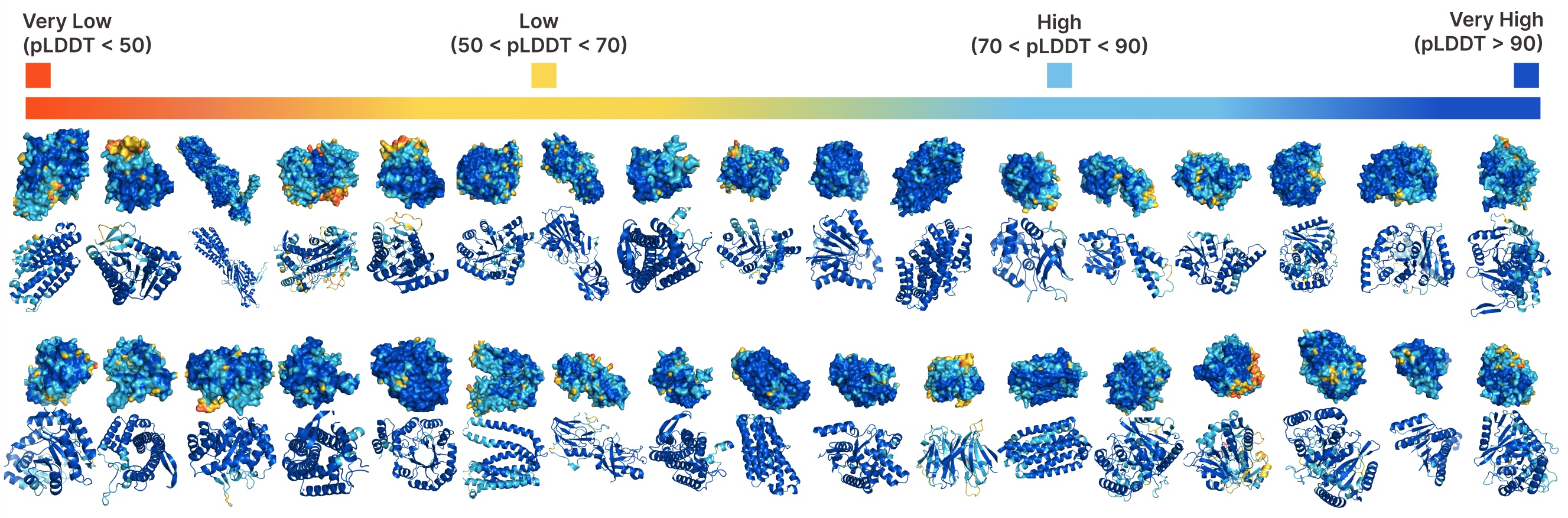 PLAIDによる無条件生成。
PLAIDによる無条件生成。
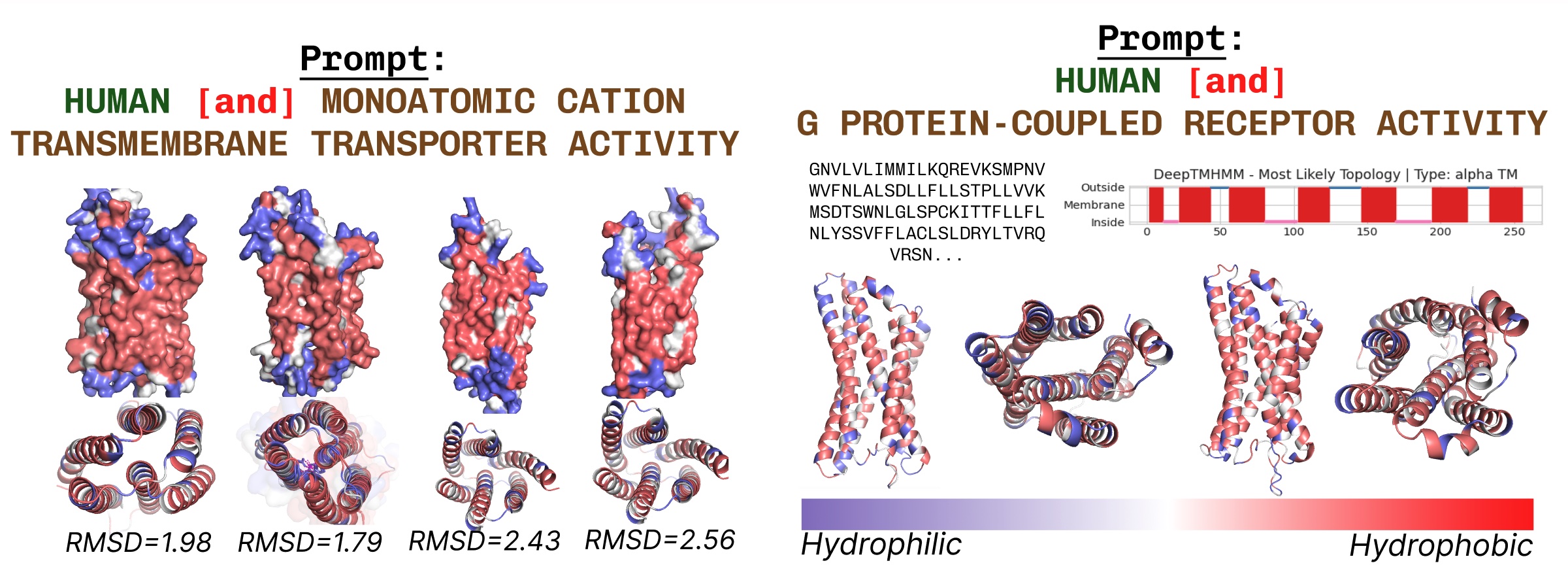 膜貫通タンパク質は、脂肪酸層に埋め込まれたコア部分に疎水性残基を持ちます。これらは、膜貫通タンパク質のキーワードでPLAIDにプロンプトを与えると一貫して観察されます。
膜貫通タンパク質は、脂肪酸層に埋め込まれたコア部分に疎水性残基を持ちます。これらは、膜貫通タンパク質のキーワードでPLAIDにプロンプトを与えると一貫して観察されます。
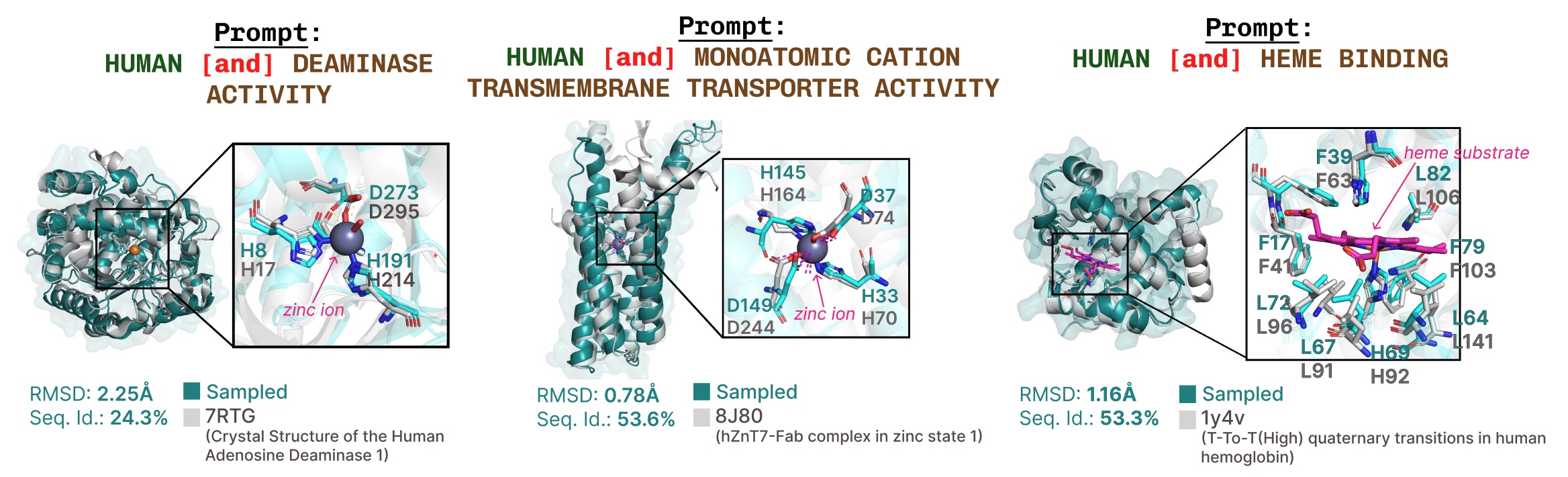 機能プロンプトに基づく活性部位再現の追加例。
機能プロンプトに基づく活性部位再現の追加例。
原文を表示
PLAID is a multimodal generative model that simultaneously generates protein 1D sequence and 3D structure, by learning the latent space of protein folding models.
The awarding of the 2024 Nobel Prize to AlphaFold2 marks an important moment of recognition for the of AI role in biology. What comes next after protein folding?
In PLAID, we develop a method that learns to sample from the latent space of protein folding models to generate new proteins. It can accept compositional function and organism prompts, and can be trained on sequence databases, which are 2-4 orders of magnitude larger than structure databases. Unlike many previous protein structure generative models, PLAID addresses the multimodal co-generation problem setting: simultaneously generating both discrete sequence and continuous all-atom structural coordinates.
From structure prediction to real-world drug design
Though recent works demonstrate promise for the ability of diffusion models to generate proteins, there still exist limitations of previous models that make them impractical for real-world applications, such as:
All-atom generation: Many existing generative models only produce the backbone atoms. To produce the all-atom structure and place the sidechain atoms, we need to know the sequence. This creates a multimodal generation problem that requires simultaneous generation of discrete and continuous modalities.
Organism specificity: Proteins biologics intended for human use need to be humanized, to avoid being destroyed by the human immune system.
Control specification: Drug discovery and putting it into the hands of patients is a complex process. How can we specify these complex constraints? For example, even after the biology is tackled, you might decide that tablets are easier to transport than vials, adding a new constraint on soluability.
Generating “useful” proteins
Simply generating proteins is not as useful as controlling the generation to get useful proteins. What might an interface for this look like?
For inspiration, let's consider how we'd control image generation via compositional textual prompts (example from Liu et al., 2022).
In PLAID, we mirror this interface for control specification. The ultimate goal is to control generation entirely via a textual interface, but here we consider compositional constraints for two axes as a proof-of-concept: function and organism:
Learning the function-structure-sequence connection. PLAID learns the tetrahedral cysteine-Fe2+/Fe3+ coordination pattern often found in metalloproteins, while maintaining high sequence-level diversity.
Training using sequence-only training data
Another important aspect of the PLAID model is that we only require sequences to train the generative model! Generative models learn the data distribution defined by its training data, and sequence databases are considerably larger than structural ones, since sequences are much cheaper to obtain than experimental structure.
Learning from a larger and broader database. The cost of obtaining protein sequences is much lower than experimentally characterizing structure, and sequence databases are 2-4 orders of magnitude larger than structural ones.
How does it work?
The reason that we’re able to train the generative model to generate structure by only using sequence data is by learning a diffusion model over the latent space of a protein folding model. Then, during inference, after sampling from this latent space of valid proteins, we can take frozen weights from the protein folding model to decode structure. Here, we use ESMFold, a successor to the AlphaFold2 model which replaces a retrieval step with a protein language model.
Our method. During training, only sequences are needed to obtain the embedding; during inference, we can decode sequence and structure from the sampled embedding. ❄️ denotes frozen weights.
In this way, we can use structural understanding information in the weights of pretrained protein folding models for the protein design task. This is analogous to how vision-language-action (VLA) models in robotics make use of priors contained in vision-language models (VLMs) trained on internet-scale data to supply perception and reasoning and understanding information.
Compressing the latent space of protein folding models
A small wrinkle with directly applying this method is that the latent space of ESMFold – indeed, the latent space of many transformer-based models – requires a lot of regularization. This space is also very large, so learning this embedding ends up mapping to high-resolution image synthesis.
To address this, we also propose CHEAP (Compressed Hourglass Embedding Adaptations of Proteins), where we learn a compression model for the joint embedding of protein sequence and structure.
Investigating the latent space. (A) When we visualize the mean value for each channel, some channels exhibit “massive activations”. (B) If we start examining the top-3 activations compared to the median value (gray), we find that this happens over many layers. (C) Massive activations have also been observed for other transformer-based models.
We find that this latent space is actually highly compressible. By doing a bit of mechanistic interpretability to better understand the base model that we are working with, we were able to create an all-atom protein generative model.
Though we examine the case of protein sequence and structure generation in this work, we can adapt this method to perform multi-modal generation for any modalities where there is a predictor from a more abundant modality to a less abundant one. As sequence-to-structure predictors for proteins are beginning to tackle increasingly complex systems (e.g. AlphaFold3 is also able to predict proteins in complex with nucleic acids and molecular ligands), it’s easy to imagine performing multimodal generation over more complex systems using the same method. If you are interested in collaborating to extend our method, or to test our method in the wet-lab, please reach out!
If you’ve found our papers useful in your research, please consider using the following BibTeX for PLAID and CHEAP:
@article{lu2024generating, title={Generating All-Atom Protein Structure from Sequence-Only Training Data}, author={Lu, Amy X and Yan, Wilson and Robinson, Sarah A and Yang, Kevin K and Gligorijevic, Vladimir and Cho, Kyunghyun and Bonneau, Richard and Abbeel, Pieter and Frey, Nathan}, journal={bioRxiv}, pages={2024--12}, year={2024}, publisher={Cold Spring Harbor Laboratory} }
@article{lu2024tokenized, title={Tokenized and Continuous Embedding Compressions of Protein Sequence and Structure}, author={Lu, Amy X and Yan, Wilson and Yang, Kevin K and Gligorijevic, Vladimir and Cho, Kyunghyun and Abbeel, Pieter and Bonneau, Richard and Frey, Nathan}, journal={bioRxiv}, pages={2024--08}, year={2024}, publisher={Cold Spring Harbor Laboratory} }
You can also checkout our preprints (PLAID, CHEAP) and codebases (PLAID, CHEAP).
Some bonus protein generation fun!
Additional function-prompted generations with PLAID.
Unconditional generation with PLAID.
Transmembrane proteins have hydrophobic residues at the core, where it is embedded within the fatty acid layer. These are consistently observed when prompting PLAID with transmembrane protein keywords.
Additional examples of active site recapitulation based on function keyword prompting.
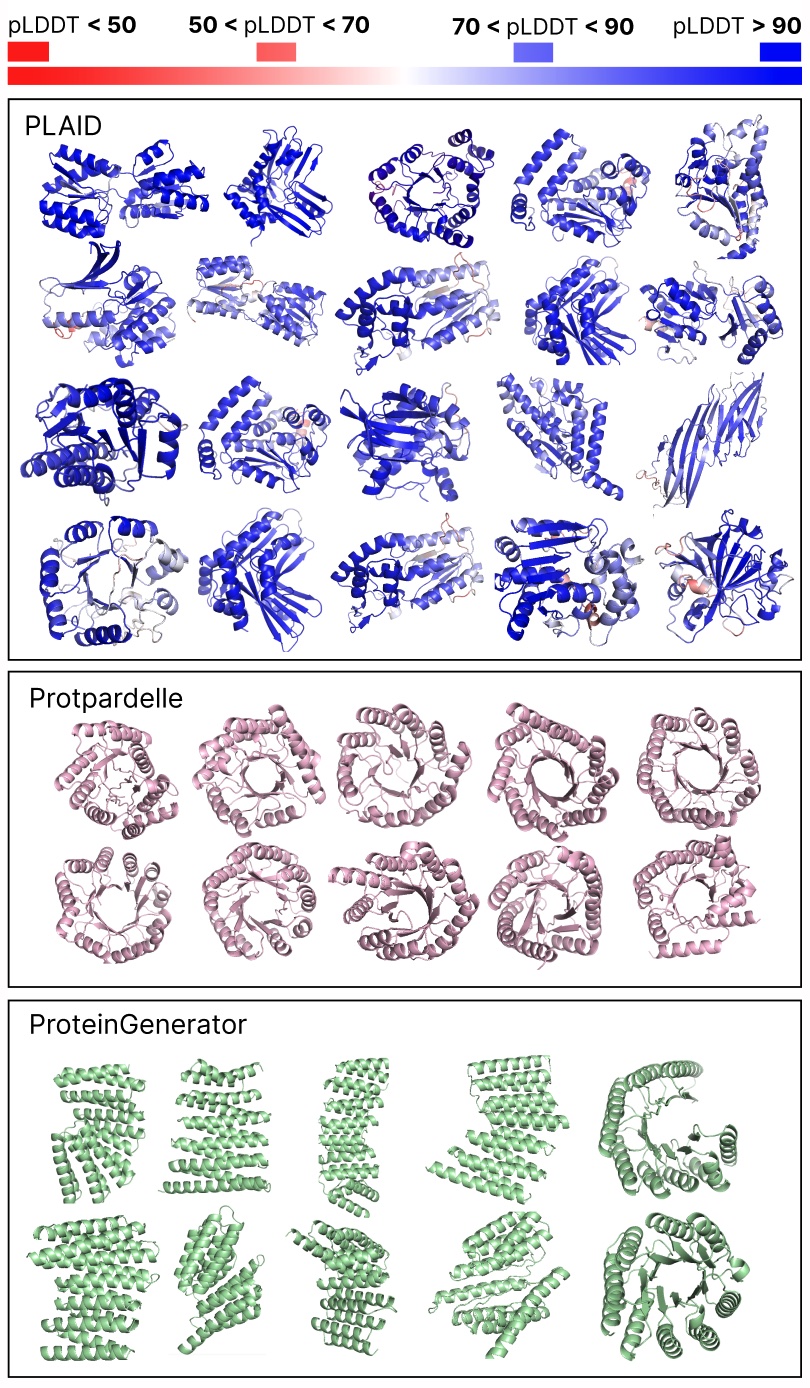 Comparing samples between PLAID and all-atom baselines. PLAID samples have better diversity and captures the beta-strand pattern that has been more difficult for protein generative models to learn.
Comparing samples between PLAID and all-atom baselines. PLAID samples have better diversity and captures the beta-strand pattern that has been more difficult for protein generative models to learn.
Acknowledgements
Thanks to Nathan Frey for detailed feedback on this article, and to co-authors across BAIR, Genentech, Microsoft Research, and New York University: Wilson Yan, Sarah A. Robinson, Simon Kelow, Kevin K. Yang, Vladimir Gligorijevic, Kyunghyun Cho, Richard Bonneau, Pieter Abbeel, and Nathan C. Frey.
関連記事
今日のまとめ
AI日報で今日の重要ニュースをまとめ読み