化学者としての Claude の活用(12 分読)
Anthropic は世界最高峰の化学者と協力し、Claude が NMR スペクトルなどの専門的な分析入力を読み解く能力を強化する取り組みを発表した。
キーポイント
多様な表現形式の翻訳課題
化学者はホワイトボードのスケッチ、機器の読み取り値、データベースクエリなど異なる形式間を行き来する必要があり、これら全てを正確に解釈・変換する能力が求められる。
AI 導入の障壁と現状
過去の AI ツールはデータ不足(特にネガティブ結果や非構造化情報の欠如)により普及が遅れていたが、最新のマルチモーダル・推論モデルがこの課題を解決する可能性を示している。
Claude の化学専門家化プロジェクト
Anthropic は合成・計算・分析化学の専門家と連携し、特に NMR スペクトル解析という頻出タスクにおいて Claude の性能を検証・向上させる具体的な取り組みを開始した。
分子認識の重要性
構造のわずかな違い(異性体など)が薬効や毒性に劇的な影響を与えるため、AI による正確な分子認識は医薬品開発や材料科学において極めて重要である。
重要な引用
Reroute a handful of bonds among the same atoms, and glucose becomes fructose... Flip a molecule into its mirror image, and a sedative becomes a teratogen
AI is well-positioned to take on this research burden, yet it still remains largely aspirational in the context of chemistry.
Today's frontier models are multimodal, and capable of explicit reasoning. They can read a chemical structure directly from a journal figure.
影響分析・編集コメントを表示
影響分析
この記事は、汎用 AI が特定の専門領域(化学)において実用的な価値を発揮し始めた転換点を示しています。特に、データ不足という従来のボトルネックを、最新の推論モデルの能力と専門家との連携によって克服しようとする姿勢は、科学技術分野における AI 応用の新たなパラダイムを示唆しており、医薬品開発や材料探索のプロセス変革に寄与する可能性があります。
編集コメント
化学という極めて専門的でデータが複雑な領域において、AI が単なるツールから「専門家パートナー」として機能し始める兆候が見られます。特に NMR 解析のような定型的かつ重要なタスクへの適用は、実用化の第一歩として注目すべき動きです。
*要約:私たちは、世界最高峰の合成化学者、計算化学者、分析化学者と協力して、Claude の化学分野での能力を向上させる取り組みを行っています。この投稿では、その一環として行われた最初の成果について共有します。Anthropic の化学者であるデイビッド・カンバーが、化学者が最も頻繁に扱う分析的入力である NMR スペクトルに対する Claude の性能を検証したものです。*分子を取り扱う際、化学者はホワイトボード上の手書き構造図、機器の読み取り値、データベース検索クエリ文字列、特許や論文における技術的表記の間を行き来します。これらすべての表現は同じ基礎となる化学をエンコードしていますが、それぞれが異なる種類の流暢さを要求します。例えば、カフェインのスケッチを見れば、化学者はそれが体内の眠気信号であるアデノシンに似ていることに気づき、受容体をブロックすることで私たちが覚醒し続けることを予測できます。しかし、その同じスケッチでは、他のほぼ同一視される分子と区別することはできません。
化学者が取り扱っている分子を理解することは極めて重要です。化学は、私たちが摂取する食品や医薬品から、ローション、塗料、プラスチックに至るまで、あらゆるものの基盤となっています。同じ原子間にあるいくつかの結合を再配置するだけで、グルコースがフルクトースに変わり、同じ化学式を持ちながら全く異なる代謝経路で処理される分子になります。分子を鏡像反転させると、鎮静剤が奇形誘発物質へと変化し、サリドマイド の悲劇のように悲惨な結果を招きます。化学者の日常業務は、与えられたタスクに適した表現形式において、これらのシグナルを正しく読み取ることに依存しています。
これらの表現間の翻訳(図から構造を追跡する、機器の読値と提案された生成物を照合する、適切な記法でデータベースを検索するなど)には時間がかかり、大規模なスケールで追いつくことは不可能です。最大の化学登録所である CAS は、すでに 2 億 9000 万件以上の公開物質をカタログ化しており、毎日約 1 万 5000 件の新しい物質が追加されています。
AI はこの研究負担を引き受けるのに適した立場にあるが、化学の文脈においては依然として主に理想論にとどまっている。機械学習ツールは長年、逆合成(標的分子からより単純な前駆体へと遡ってその構築方法を計画するプロセス)や反応予測、物性推定において変革をもたらすものとして位置づけられてきたが、これらのツールが必要とするデータは入手困難であり、結果のないデータが不足しており、フォーマットに一貫性がなく、購読ジャーナルの有料壁(および非構造化された補足情報)の向こう側に閉じ込められている。逆合成はその典型例である——能力のある AI ツールは何年も存在してきたが、その普及は偏っており、平均的な学術研究者や小規模ラボの化学者がまだそれらを使用していない。
それでもなお、AI の進展がついに化学に到達しつつある。今日の最先端モデルはマルチモーダルであり、明示的な推論能力を備えている。これらは事前にキュレーションされた分子データベースに依存するのではなく、ジャーナルの図や手書きスケッチから直接化学構造を読み取ることができる。また、方法セクションや補足情報の実験詳細も、実際に公開されている形式のまま読み取ることもできる。さらに、推論プロセスを段階的に示すことができるため、化学者が出力を検証することも可能になる。これらはいずれも、長年この分野が指摘してきたデータの問題を解消するものではないが、それにもかかわらず解決可能な問題の範囲を変えるものである。
究極的に、私たちの主張は控えめなものです:Claude は、化学者の判断を補完する日常的な翻訳、想起、統合作業において、意味のある支援を開始しており、私たちはその有用性をさらに拡張していく計画です。今日、私たちはこの取り組みを加速させるための最初のホワイトペーパーを発表します。これは、化学者が最も頻繁に扱う分析入力である NMR スペクトルに取り組むものです。
Claude と ChemDraw の NMR 予測および構造決定における比較
**
完全版は こちら でご覧いただけます。**
薬物、農薬、染料、香料、ポリマー、DNA またはタンパク質のサブユニット、および機能的な無機物や固体材料など、ほぼすべての小分子は、化学者がその構造を決定したからこそ存在しています。これらの分子は顕微鏡では見ることができないため、化学者はスペクトル分析に頼らざるを得ません。光、電波、または磁場を用いて分子をプローブすることで、特定の分子がこのエネルギーをどのように吸収し、放出し、あるいは偏向するかというパターン、すなわちスペクトルが得られ、それによって化学者はその構造を決定することができます。
NMR 分光法は、化学者がこれに依存する代表的な手法の一つですが、合成化学における最も時間のかかる工程の一つです。化合物ごとに、化学者は手作業でスペクトル上の各ピークを提案された構造内の原子と対応付けなければなりません。このホワイトペーパーでは、Claude が今日化学者が依存している専用 NMR ソフトウェアに対してどのように振る舞うかをテストしました。Claude の 3 つのモデル(Opus 4.7, Opus 4.6, Sonnet 4.6)を、ChemDraw および MestReNova と比較し、トレーニングカットオフ後に公開された合成化学のプレプリントから抽出した 20 種類の化合物を用いて評価を行いました。これにより選択バイアスを回避しています。ChemDraw と MestReNova はどちらも順方向予測を行い、描画された構造を用いて生成される NMR スペクトルをシミュレーションします。順方向予測に加えて、Claude が逆方向の処理も可能か、つまり実験スペクトルから出発して背後にある構造を提案できるかどうかを確認したかったのです。これはより困難なタスクであり、現在既存のソフトウェアでは化学者に任されている作業です。
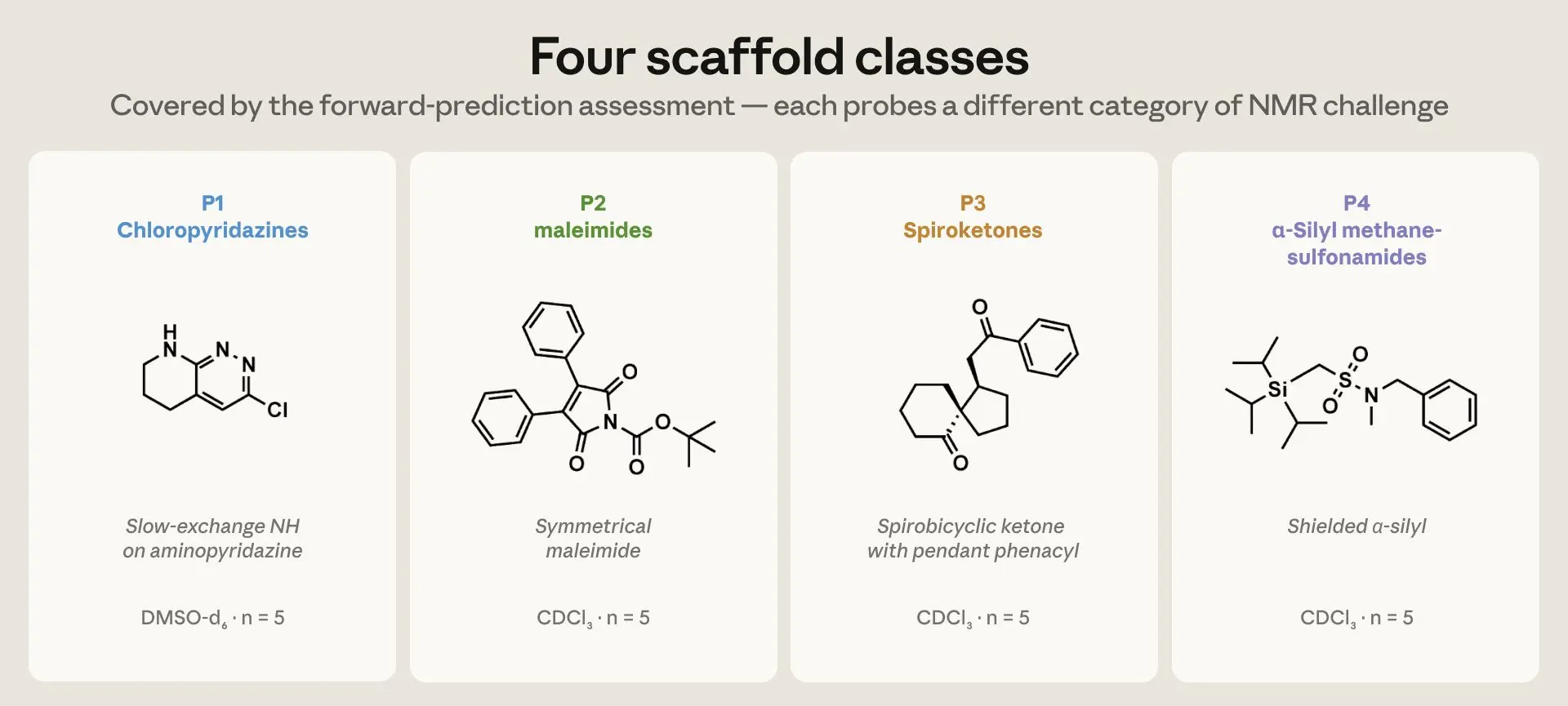
評価設定を行うために、モデルの学習カットオフ後に投稿された ChemRxiv のプレプリントから 20 種類の化合物を抽出し、各論文から初めて完全に特徴付けられた新規分子を 1 つずつ選びました。これら 20 種類は 4 つの構造ファミリーに分類され、各ファミリーには 5 種類ずつ含まれており、それぞれが異なる NMR(核磁気共鳴)の課題に関連するカテゴリーとして選定されています。各ツールには、化学者がソフトウェアに入力するために使用するテキスト記法である SMILES 文字列として符号化された構造が与えられ、1D NMR スペクトル上(ppm:百万分率で化学シフトを測定する水平軸)において、すべての水素と炭素のピークがどこに現れるかを予測するように求められました。NMR サンプルは液体に溶解されており、溶媒(クロロホルム、DMSO など)の選択によってピーク位置がわずかに変動するため、各ツールには、化学者が公開論文で使用した任意の溶媒におけるスペクトルを予測するよう指示されました。
 image*図 1. 順方向予測評価でカバーされた 4 つの骨格クラス。それぞれが異なる NMR(核磁気共鳴)課題のカテゴリーをプローブします。P1 クロロピリダジン類は、DMSO-d₆ 中でアミノピリダジンのスロー・エクスチェンジ NH を有しています;P2 Boc-N-アリルマレイミドおよび N-Boc イナミドは、α-ビニルイミドカルボニルと稀なイナミドのα/β炭素対を課題としています;P3 スピロケトンは、フェナシルまたはアセチル側鎖を持ちジアステレオトピック CH₂ を有するスピロ二環式ケトンです;P4 α-シリルメタンスルホナミドは、遮蔽されたシリコンα炭素を有しています。各クラスあたり 5 化合物、合計 n = 20 です。*
image*図 1. 順方向予測評価でカバーされた 4 つの骨格クラス。それぞれが異なる NMR(核磁気共鳴)課題のカテゴリーをプローブします。P1 クロロピリダジン類は、DMSO-d₆ 中でアミノピリダジンのスロー・エクスチェンジ NH を有しています;P2 Boc-N-アリルマレイミドおよび N-Boc イナミドは、α-ビニルイミドカルボニルと稀なイナミドのα/β炭素対を課題としています;P3 スピロケトンは、フェナシルまたはアセチル側鎖を持ちジアステレオトピック CH₂ を有するスピロ二環式ケトンです;P4 α-シリルメタンスルホナミドは、遮蔽されたシリコンα炭素を有しています。各クラスあたり 5 化合物、合計 n = 20 です。*
言語モデルの出力は実行ごとに異なるため、各 Claude モデルは化合物ごとに 3 回クエリを実行して平均化しました。一方、ChemDraw と MestReNova は毎回同じ回答を返すため、1 回のみ実行しました。その後、予測されたピークを実験値とペアリングし、ppm(化学シフト)単位での差を測定しました。その結果は、化学者が「正しい」と判断する範囲内—水素の場合は±0.20 ppm、炭素の場合は±1.0 ppm—に収まりました。
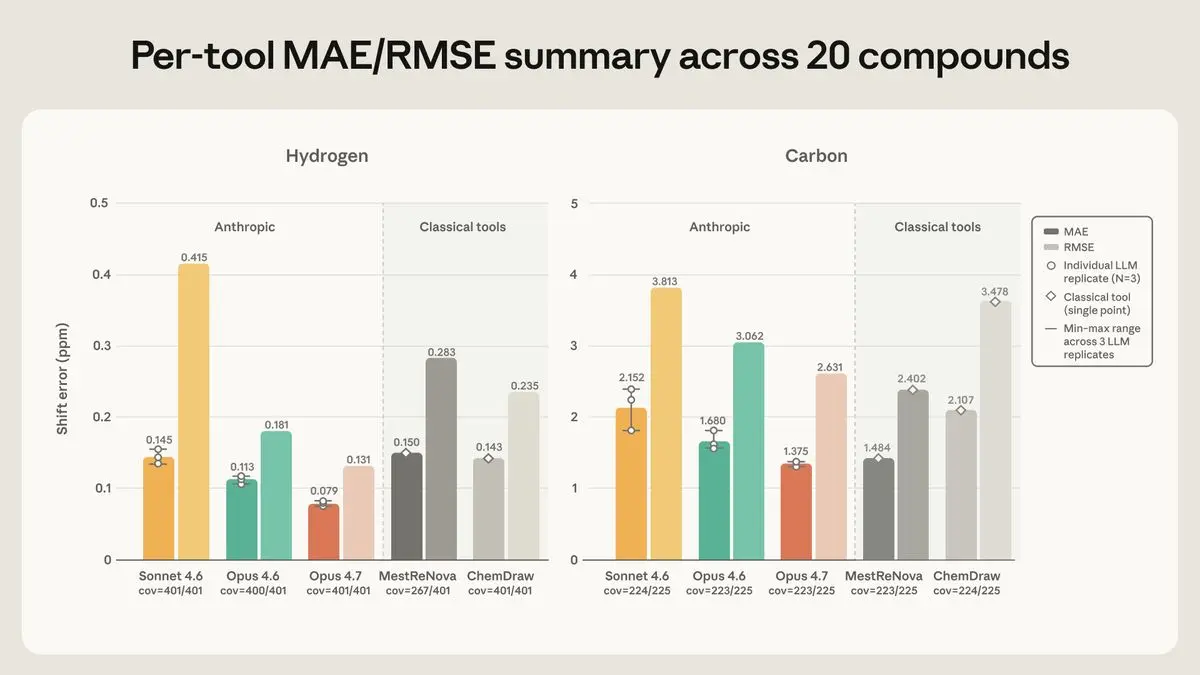
図2。20化合物における順方向予測の¹H(左)と¹³C(右)シフト誤差に対するツール別MAE(濃い色)およびRMSE(薄い色)。各ツールの下部にはカバレッジを示す。Claude の棒グラフは3回の反復実験の平均値に最小値~最大値の範囲を示し、重ねて反復実験の点をプロットしたものである。古典的ツールは単一ポイント予測であり(範囲なし)。
水素については、Opus 4.7 が最も精度が高く、平均誤差は±0.079 ppm で、許容幅の半分以下に収まり、ピークがその範囲内に収まる割合も最高であった。炭素については、Opus 4.7 と MestReNova がほぼ同点で、それぞれ±1.37 ppm および±1.48 ppm であり、残りのツールは両元素において同じ順位付けを維持した。Opus 4.6 は予想通り中程度であり、Sonnet 4.6 が最も弱かった。この差が最も顕著だったのは、 notoriously difficult(非常に困難な)水素原子の一つ、すなわちクロロピリダジン族における NH プロトンである。その真の位置は 6.8~7.9 ppm の狭い帯域に存在する。Opus 4.7 はこれをわずかに低めに、かつ一貫して予測した。一方、Opus 4.6 は推測値を数 ppm にわたってばらつかせ、Sonnet 4.6 は 10~13 ppm の範囲に置いたが、これは実際の出現位置から大きく外れている。
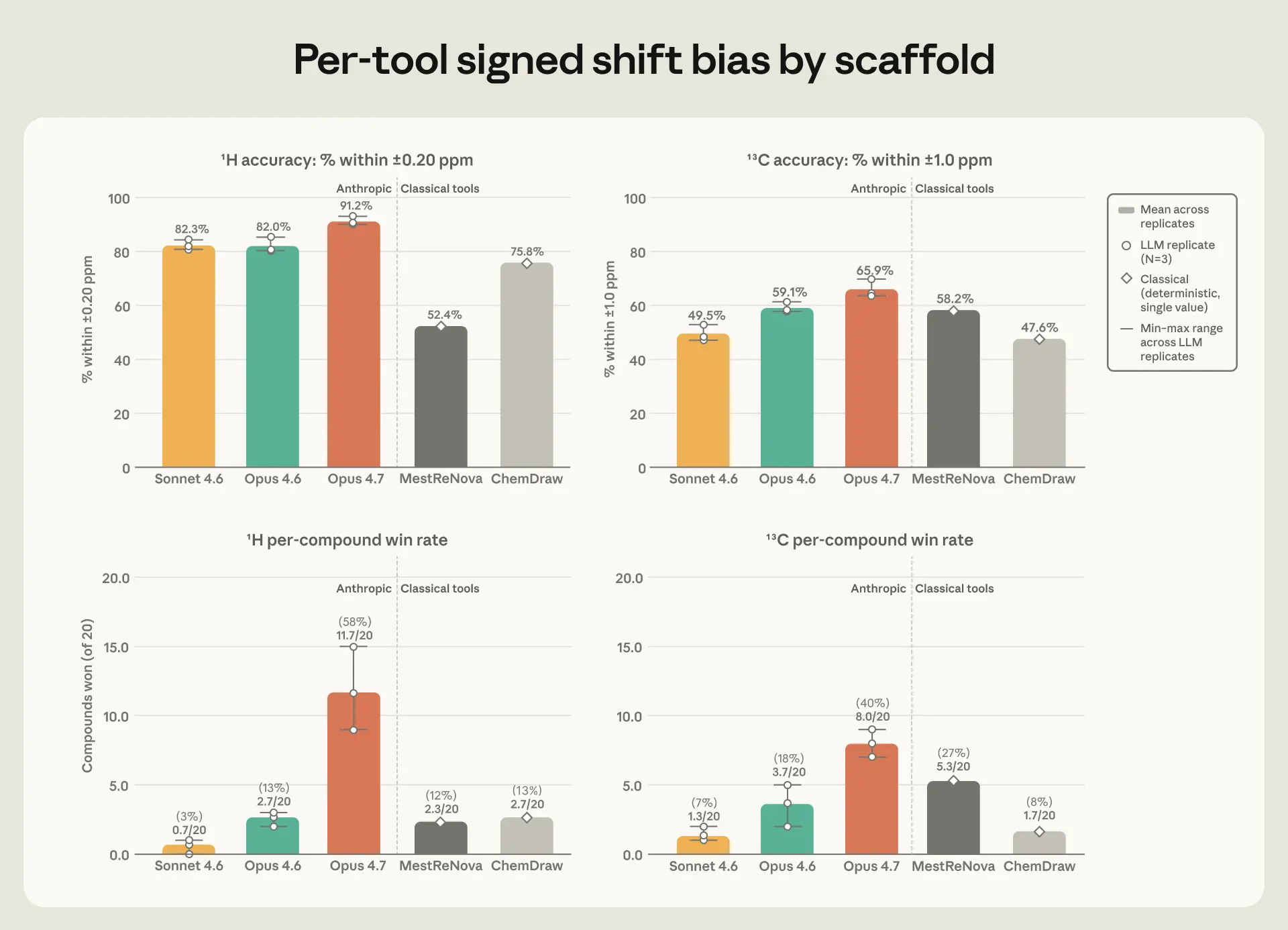
 image*図 3. 上部:実験値との原子位置の一致率(±0.20 ppm 以内、¹H 左側および ±1.0 ppm 以内、¹³C 右側)。下部:化合物ごとの勝率(ツールが 20 化合物中、各化合物の平均二乗誤差 MAE が最小となった化合物の割合)。Claude の棒グラフは 3 回の反復実験の平均値と最小値〜最大値の範囲を示す;古典的ツールは単一点予測に基づくもの。*
image*図 3. 上部:実験値との原子位置の一致率(±0.20 ppm 以内、¹H 左側および ±1.0 ppm 以内、¹³C 右側)。下部:化合物ごとの勝率(ツールが 20 化合物中、各化合物の平均二乗誤差 MAE が最小となった化合物の割合)。Claude の棒グラフは 3 回の反復実験の平均値と最小値〜最大値の範囲を示す;古典的ツールは単一点予測に基づくもの。*
Opus 4.7 は ChemDraw や MestReNova と比較して概ね同等の性能を発揮しましたが、水素原子の NMR(核磁気共鳴スペクトル)ピークの形状や、ピーク間の距離を予測する点では差が広がりました。これらの特徴は、化学者が位置情報とともに読み取る構造情報も含まれています。Opus 4.7 は、実験的に報告された分裂パターンを他のどのツールよりも頻繁に再現しました。また、Claude モデル 3 つはいずれも、約 80% のケースでサブピーク間の間隔を半ヘルツ以内の精度で予測しました。一方、ChemDraw と MestReNova ではその割合は 26〜35% に留まりました。さらに、Opus 4.7 は 3 回の反復実験全体を通じて最も一貫した結果を示し、その平均誤差が実験ごとに変動する幅は、次点のツールとの差よりも小さかったのです。
そこから、逆予測(構造決定)を評価しました。すなわち、分子のスペクトルからその構造を特定できるかという問いです。Opus 4.7 に構造決定問題 15 問を与え、それぞれ 3 回ずつ、上位 3 つの候補構造を提案するよう依頼しました。各問題には、化合物の正確な分子式(高分解能質量分析から得られたもの)と、水素・炭素 NMR スペクトルが提供されました。15 問は難易度別に分割されています。8 つの比較的簡単なターゲット(単環または 2 断片からなる分子)には、分子式とスペクトルのみを与えました。一方、7 つのより複雑なターゲット(縮合環、スピロ環など)には、反応に用いられた出発物質の構造という追加の手がかりも併せて提示しました。
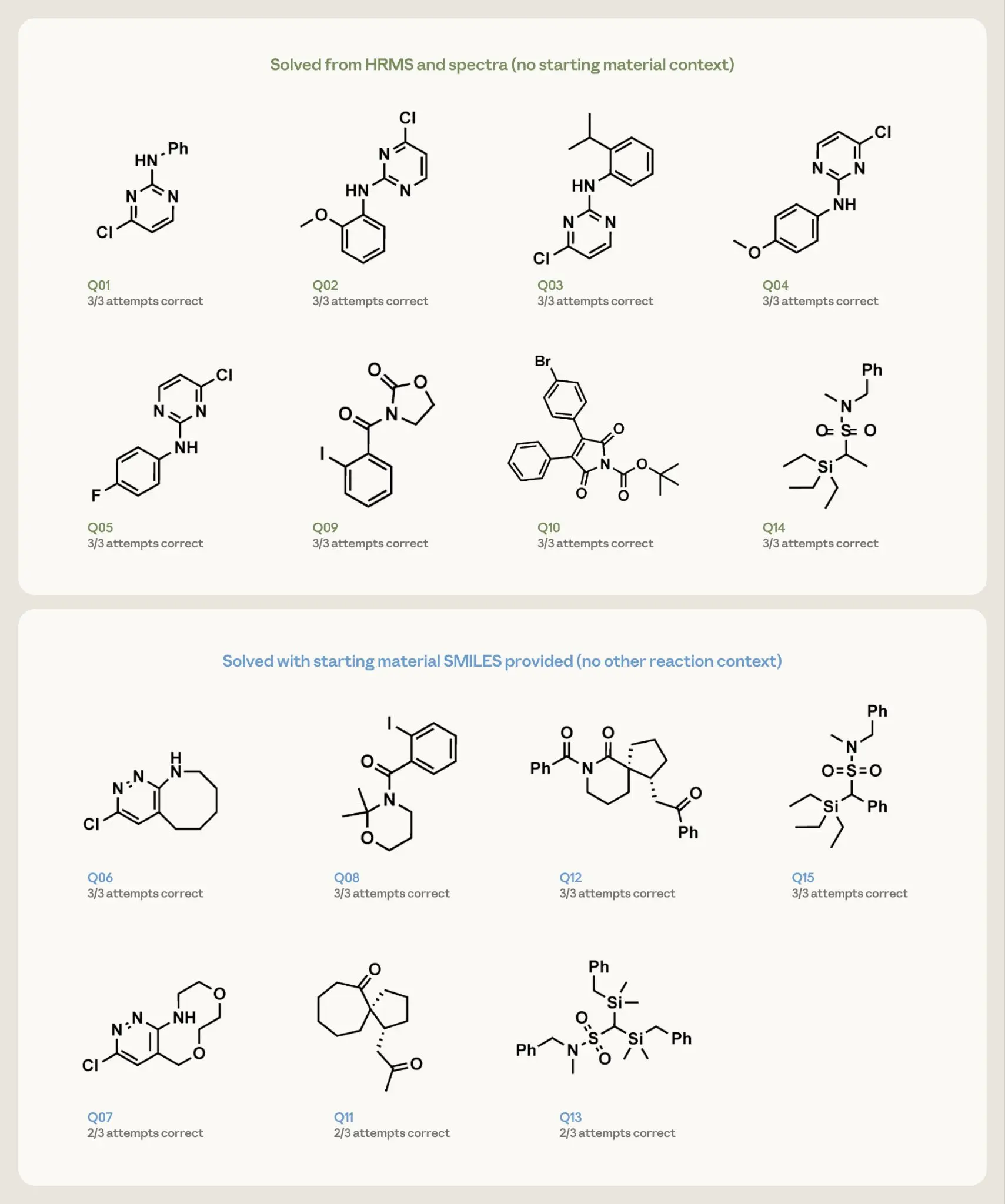 image*図 4. 15 の逆タスク問題における構造決定の結果。各パネルは、公開されたターゲットとその 3 回の試行での成功回数を示しています。枠線の色はプロンプト条件を示します:緑はスペクトルと高分解能質量分析(HRMS)のみで、出発物質の文脈なし;青はスペクトル、HRMS、および出発物質の SMILES を含み、他の反応文脈なし。
image*図 4. 15 の逆タスク問題における構造決定の結果。各パネルは、公開されたターゲットとその 3 回の試行での成功回数を示しています。枠線の色はプロンプト条件を示します:緑はスペクトルと高分解能質量分析(HRMS)のみで、出発物質の文脈なし;青はスペクトル、HRMS、および出発物質の SMILES を含み、他の反応文脈なし。
Opus 4.7 は、スペクトルと分子式のみから、すべての試行において8つのより単純な構造をすべて回復しました。7 つのより困難なターゲットについては、出発物質の手がかりを与えられた場合、そのうち4 つでは3 回の試行すべてで正しい構造を返すことに成功し、残りのものについても3 回中2 回の試行で正解を導き出しました。
最終的に、日常的なデータ予測においては、化学分野に特化したファインチューニングを行っていない汎用モデルである Opus 4.7 が、平均的には ChemDraw や MestReNova と同等かそれ以上に優れていることがわかりました。さらに、Claude は問題の逆方向にも処理でき、NMR データのみから構造を提案することも可能です。構造決定のための専用ソフトウェアは数十年にわたり存在していますが、通常は 2D NMR(2 つの軸を持つスペクトルであり、出力はピークの列ではなく等高線マップとなる)や、専門的なトレーニング、ライセンスが必要なツールを必要とします。一方、Claude は化学者がチャットに貼り付けるような高解像度質量スペクトルと 1D ピークリストのみから、セットアップ不要でこれを実現します。
Limitations
この評価は、汎用モデルが NMR ソフトウェアと競合しうることを示しており、さらに 1D 逆方向の構造決定を扱いやすくできる可能性も示しています。しかし、いくつかの注目すべき限界点もあります。
- まず、評価対象は小規模でした。順方向タスクでは4つの骨格構造にまたがる20化合物、逆方向タスクでは15化合物であり、各骨格構造は単一の失敗モードのクラスに寄与しています。したがって、モデルのパフォーマンスは精密な数値というよりは示唆的な指標として読むべきです。
- 第二に、最も密度の高い逆方向ターゲットにおいて、出発物質を追加入力として用いない場合、モデルは最終構造を確定せずに推論ループに陥ることがあります。そのため、7つのより困難な問題では、スペクトル単独ではなく、出発物質の構造情報を含めて提示されました。
- 第三に、一部の化学骨格構造は未検証のままです。例えば、溶媒との交換が遅く鋭いNMRピークを残すような緩徐交換型NHヘテロ芳香族化合物(aromatic rings whose N–H exchanges with solvent slowly enough to leave a sharp NMR peak)は、クロロピリダジンを通じてのみサンプリングされており、関連する系(ヒドロキシピリジン、アミノチアゾール、および他のDMSO-d₆でNH活性を示す骨格構造)は除外されています。
- 第四に、2D実験(COSY, HSQC, HMBC)と立体化学は、1D NMRのみでは配置を決定できないため、設計上対象外となっています。その結果、複雑な天然物化合物の評価は行われませんでした。
- 最後に、溶媒のカバレッジはDMSO-d₆、CDCl₃、およびD₂Oに限定されており、メタノール-d₄、ベンゼン-d₆、アセトン-d₆については評価されていません。
理想的には、20〜30の骨格構造クラスにまたがる数百化合物、各クラスあたり少なくとも15化合物を用いてこれらの数値がどの程度維持されるかを確認したいところです。これにより、クラス内変動とツール間の差異を分離できます。また、クロロピリダジン以外のNH活性ヘテロ芳香族化合物の評価、未検証溶媒の検討、および2D実験を活用した両タスクのバージョン実施も行うべきでしょう。
先を見据えて
Claude の化学分野におけるパフォーマンスを継続して向上させるにあたり、私たちは特に化学者の作業を最も阻害するいくつかのボトルネックに焦点を当てています。
- 化学構造の読み取りとレンダリング:図版、特許、スライド、スケッチからの描画を機械可読形式に変換すること、および化学文献で用いられる体系的名称との間で構造表現を行き来すること。
- 反応および合成推論:合成経路の提案・評価・批判、結果の予測、選択性や条件、および生成されやすい副生成物についての検討。
- 反応機構:電子矢印、中間体、遷移状態に関する議論を含む、化学者が実際に使用する言語で反応機構を説明し検証すること。
- 化学文献の理解:出版された研究において現れる化学を解釈すること。同じ分子が描画されたり名称付けられたり、略称されたりコードで参照されたりする中で、方法論セクション、補足情報、特許から重要な化学情報を抽出すること。
これらはすべて同じ成熟度曲線上にあるわけではありません。スペクトル解析はベンチマーク可能な段階まで進んでいる一方で、逆合成計画のような他の分野はまだ範囲を定義している最中です。これらのボトルネックに対する理解が深まるにつれ、現在のモデルがどこで優れているか、またどこでまだ不足しているかを共有していく予定です。私たちの究極の目標は、現場の化学者が Claude によって時間を節約できる箇所と、依然として自身の専門知識に頼る必要がある箇所を明確に認識できるようにすることです。
私たちと一緒に働く
私たちは、化学研究をより明確に支援するために AI for Science program を拡大しています。Claude が実際に役立つ可能性のある問題に取り組んでいる研究者、特に私たちが説明したような多モーダル推論(multimodal reasoning)を要する課題に取り組んでいる方々には、scienceblog@anthropic.com まで、または AI for Science の申請フォームを通じてご連絡ください。
脚注
- 朝吐き気止め薬が世界中で 10,000 人以上の子供に深刻な先天異常を引き起こしたと関連付けられた事件。
- 私たちが化合物を抽出した 4 つのプレプリント(preprint): https://chemrxiv.org/doi/full/10.26434/chemrxiv.15002274/v1, https://chemrxiv.org/doi/full/10.26434/chemrxiv-2025-59lfh, https://chemrxiv.org/doi/full/10.26434/chemrxiv.15002423/v1, https://chemrxiv.org/doi/full/10.26434/chemrxiv.15002316/v1。
関連コンテンツ
社会科学におけるコーディングエージェント
AI およびコーディングエージェントの使用に関する社会学者 1,260 名を対象とした調査の結果。
Project Glasswing: 初期アップデート
Project Glasswing から得た知見についての初期レポートです。
2028 年:世界の AI リーダーシップに関する 2 つのシナリオ
米国と中国間の AI 競争に関する私たちの見解。
原文を表示
*Summary: We’re working with world-class synthetic, computational, and analytical chemists to make Claude better at chemistry. In this post, we share our first work as part of this effort, in which Anthropic chemist, David Kamber, examines how Claude performs on a chemist’s most common analytical input, an NMR spectrum.*When working with molecules, chemists move between hand-drawn structures on a whiteboard, instrument readouts, database query strings, and the technical notations of patents and publications. Each of these representations encodes the same underlying chemistry, but each demands a different kind of fluency. A sketch of caffeine, for example, allows a chemist to spot its resemblance to adenosine, the body’s drowsiness signal, and predict that it keeps us alert by blocking the receptor. However, that same sketch cannot help a chemist tell it apart from other near-identical looking molecules.
Understanding what molecule a chemist is working with is critical. Chemistry undergirds everything from the foods and medicine we ingest to our lotions, paints, and plastics. Reroute a handful of bonds among the same atoms, and glucose becomes fructose, molecules sharing a formula but processed through entirely different metabolic pathways. Flip a molecule into its mirror image, and a sedative becomes a teratogen, as happened in the thalidomide disaster.1 Chemists’ everyday work depends on reading these signals correctly across whichever representation befits a given task.
Translating between these representations (chasing down a structure from a figure, reconciling an instrument readout against a proposed product, querying a database in the right notation) is time consuming and impossible to keep up with at scale—CAS, the largest chemistry registry, catalogs over 290 million disclosed substances and grows by roughly 15,000 new ones every day.
AI is well-positioned to take on this research burden, yet it still remains largely aspirational in the context of chemistry. Machine-learning tools have been positioned for years as transformative for retrosynthesis—the process of working backward from a target molecule to simpler precursors to plan how to build it—reaction prediction, and property estimation, but the data those tools need have been hard to come by—sparse on null-results, inconsistent in format, and locked behind paywalls at subscription journals (and in unstructured supporting information). Retrosynthesis is a case in point—capable AI tools have existed for years, but adoption is uneven, and the average academic or small-lab chemist still doesn't use them.
Even so, advancements in AI are finally reaching chemistry. Today’s frontier models are multimodal, and capable of explicit reasoning. They can read a chemical structure directly from a journal figure or hand sketch rather than depending on a pre-curated molecular database. And they can read the experimental detail of a methods section or supporting information in the form it is actually published. They can also show their reasoning step by step, which means a chemist can audit the outputs. None of this eliminates the data problem the field has been describing for years, but it changes which problems are tractable despite it.
Ultimately, our claim is a modest one: Claude is starting to meaningfully assist chemists with the daily translation, recall, and integration work that complements their judgment, and we plan to keep extending its helpfulness. Today we are publishing the first white paper in the effort to accelerate this work. It tackles a chemist's most common analytical input: an NMR spectrum.
Claude vs. ChemDraw on NMR prediction and structure elucidation
**
Full version can be found here**
Nearly every small molecule—drug, pesticide, dye, fragrance, polymer, DNA or protein subunit, and functional inorganic or solid-state material—exists because a chemist determined its structure. Given that these molecules cannot be seen with microscopes, chemists must rely on spectral analysis, probing a molecule with light, radio waves, or magnetic fields. The way a given molecule absorbs, emits, or deflects this energy gives chemists a pattern, or spectrum, with which they can elucidate its structure.
NMR spectroscopy—one of the canonical techniques chemists rely on for this—is one of the most time-consuming steps in synthetic chemistry; for every compound, a chemist has to match each peak in the spectrum to an atom in the proposed structure by hand. For this white paper, we tested how Claude fared against the dedicated NMR software chemists rely on today. We measured three Claude models (Opus 4.7, Opus 4.6, Sonnet 4.6) against ChemDraw and MestReNova on 20 compounds drawn from synthetic chemistry preprints published after the models’ training cutoff so as to avoid selection bias. Both ChemDraw and MestReNova do forward prediction, using a drawn structure to simulate what NMR spectrum will be produced. In addition to forward prediction, we also wanted to see whether Claude could go the other direction—starting from an experimental spectrum and proposing the structure behind it. This is the harder task, and the one existing software currently leaves to the chemist.
To set up our assessment, we pulled 20 compounds from ChemRxiv preprints2 posted after the models’ training cutoff, taking the first fully characterized novel molecules from each paper. The 20 span four structural families, five compounds each, with each family selected because it involves a different category of NMR challenge. Each tool was given the structure encoded as a SMILES string—the line-of-text notation chemists use to input a molecule to software—and was asked to predict where every hydrogen and carbon peak would fall along a 1D NMR spectrum (a horizontal axis measuring chemical shifts in ppm, parts per million). Given that NMR samples are dissolved in a liquid, and that the choice of solvent (chloroform, DMSO, etc.) moves the peak positions slightly, each tool was told to predict the spectrum in whatever solvent the chemists had used in the published paper.
Because a language model’s output varies between runs, each Claude model was queried three times per compound and averaged; ChemDraw and MestReNova return the same answer every time and were run once. We then paired each predicted peak with its experimental counterpart and measured the gap in ppm. These landed within the window a chemist would call correct—±0.20 ppm for hydrogen or ±1.0 ppm for carbon.
On hydrogen, Opus 4.7 was most accurate, with an average error of ±0.079 ppm—well under half the tolerance window—and the highest share of peaks landing inside it. On carbon, Opus 4.7 and MestReNova were effectively tied, at ±1.37 and ±1.48 ppm; the remaining tools kept the same rank order on both elements. Opus 4.6 was predictably middling, and Sonnet 4.6 was the weakest. The gap between them was most evident on a single notoriously difficult hydrogen—an NH proton in the chloropyridazine family whose true position falls in a narrow band between 6.8 and 7.9 ppm. Opus 4.7 placed it slightly low but consistently so; Opus 4.6 scattered its guesses across several ppm; Sonnet 4.6 put it in the 10–13 range, well outside where it actually appears.
While Opus 4.7 performed fairly comparably to ChemDraw and MestReNova, the gap was wider on predicting the shape taken by a hydrogen’s NMR peak and how far apart the peaks sit, features which also contain structural information a chemist reads alongside position. Opus 4.7 matched the experimentally reported splitting pattern more often than any other tool, and all three Claude models predicted the sub-peak spacing to within half a hertz roughly 80% of the time—against 26 to 35% for ChemDraw and MestReNova. Opus 4.7 was also the most consistent across its three repeat runs: its average error varied less from run to run than the margin separating it from the next-best tool.
From there, we evaluated inverse prediction (structure elucidation): could we determine the structure of a molecule from its spectrum? We gave Opus 4.7 15 elucidation problems and asked it, three times each, to propose up to three ranked candidate structures. Each supplied the compound’s exact molecular formula (from high-resolution mass spectrometry) and its hydrogen and carbon NMR spectra. The fifteen were split by difficulty. The eight simpler targets—single-ring or two-fragment molecules—were posed with only the formula and spectra. The seven denser targets—fused rings, spirocycles, and similar—were accompanied by one additional hint: the structure of the starting material that had gone into the reaction.
Opus 4.7 recovered all eight simpler structures on every attempt from spectra and formula alone. On the seven harder targets, given the starting-material hint, it returned the correct structure on all three runs for four of them and on two of three runs for those that remained.
Ultimately, we found that for routine data prediction Opus 4.7—a general-purpose model without chemistry-specific fine-tuning—is now as good as or better than ChemDraw and MestReNova on average. Additionally, Claude can also work the problem in reverse, proposing a structure from NMR data alone. Dedicated structure-elucidation software has existed for decades, but it typically requires 2D NMR (a spectrum with two axes, and the output is a contour map rather than a row of peaks), specialized training, and licensed tools. Claude does it from the same high-resolution mass spectrum and 1D peak list a chemist would paste into a chat, with no setup.
Limitations
This assessment shows us that a general-purpose model can be competitive with NMR software and even make 1D inverse elucidation tractable. But there are a handful of noteworthy limitations.
- First, the evaluation was small—20 compounds across four scaffolds for the forward task, 15 for the inverse task—and each scaffold contributes a single class of failure modes. The model performance should thus be read as indicative rather than precise.
- Second, on the densest inverse targets, without the starting material as an additional input, the model could loop through its reasoning without committing to a final structure; this is why the seven harder problems were posed with the starting-material structure rather than spectra alone.
- Third, some chemical scaffolds were left untested. For example, slow-exchange NH heteroaromatics (aromatic rings whose N–H exchanges with solvent slowly enough to leave a sharp NMR peak) are sampled only through chloropyridazines, leaving out related systems (hydroxypyridines, aminothiazoles, and other DMSO-d₆ NH-active scaffolds).
- Fourth, 2D experiments (COSY, HSQC, HMBC) and stereochemistry are out of scope by design, since 1D NMR alone cannot fix configuration. As a result, complex natural product compounds were not evaluated.
- And finally, our solvent coverage was limited to DMSO-d₆, CDCl₃, and D₂O, so methanol-d₄, benzene-d₆, and acetone-d₆ are not assessed.
Ideally, we would see how these numbers hold up across several hundred compounds spanning 20–30 scaffold classes, with at least 15 compounds per class so that within-class variance can be separated from between-tool differences. We would also evaluate NH-active heteroaromatics beyond chloropyridazines, assess the untested solvents, and conduct versions of both tasks that draw on 2D experiments.
Looking ahead
As we continue to improve Claude’s performance in chemistry, we are focusing specifically on a handful of bottlenecks that slow chemists down the most.
- Reading and rendering chemical structures—converting a drawing from a figure, patent, slide, or sketch into a machine-readable form, and going between structural representations and the systematic names used in chemistry literature.
- Reaction and synthetic reasoning—proposing, evaluating, and critiquing synthetic routes, anticipating outcomes, and thinking through selectivity, conditions, and likely byproducts.
- Mechanism—explaining and testing reaction mechanisms in the language a chemist actually uses, with electron arrows, intermediates, and transition-state arguments.
- Chemical literature understanding—reading chemistry as it appears in published work, where the same molecule may be drawn, named, abbreviated, or referenced by a code, and pulling out the chemistry that matters from method sections, supporting information, and patents.
These are not all on the same maturity curve. Where spectral analysis is far enough along to benchmark, others, like retrosynthesis planning, are still being scoped. As we get a better understanding of these bottlenecks, we will share where current models excel, and where they still fall short. Our ultimate goal is to ensure that working chemists know where Claude can save them time and where they still need to rely on their own expertise.
Working with us
We are expanding the AI for Science program to more explicitly support chemistry research. If you are a researcher working on a problem where Claude could plausibly help, especially one that involves the kinds of multimodal reasoning we have described, we would like to hear from you at scienceblog@anthropic.com, or through the AI for Science application.
Footnotes
- An incident in which a morning sickness medication was linked to severe birth defects in over 10,000 children worldwide.
- The four preprints from which we pulled the compounds: https://chemrxiv.org/doi/full/10.26434/chemrxiv.15002274/v1, https://chemrxiv.org/doi/full/10.26434/chemrxiv-2025-59lfh, https://chemrxiv.org/doi/full/10.26434/chemrxiv.15002423/v1, https://chemrxiv.org/doi/full/10.26434/chemrxiv.15002316/v1.
Related content
Coding agents in the social sciences
Results from a survey of 1,260 social scientists about AI and coding agent use.
Project Glasswing: An initial update
An early update on what we've learned from Project Glasswing.
2028: Two scenarios for global AI leadership
Our views on the AI competition between the US and China.
関連記事
今日のまとめ
AI日報で今日の重要ニュースをまとめ読み