ESMFold2:タンパク質にも「苦い教訓」が訪れつつある - Alex Rives氏(BioHub)
BioHub の Alex Rives は、計算リソースのスケールに依存する単純なトランスフォーマーモデル(ESMFold2)が、AlphaFold3 に匹敵あるいは凌駕する性能を示すことを発表し、タンパク質生物学における「苦い教訓」の実現を宣言した。
キーポイント
計算リソースのスケールによる性能向上
ESMFold2 は、複雑なドメイン知識を組み込むのではなく、大規模で多様なデータセットを用いた単純な BERT 型トランスフォーマーを訓練することで、AlphaFold3 に匹敵する結果を達成し、計算リソースの投入が予測可能に性能向上をもたらすことを示した。
抗体やがん治療への応用実績
Cryo-EM データを活用した ESMFold2 は、特に抗体のようなタンパク質相互作用において最先端の性能を発揮し、がんおよび免疫学における 5 つの対象で推論時間のスケーリングが機能している証拠を示した。
大規模タンパク質アトラスの公開
ESMFold2 の発表に伴い、68 億個のタンパク質と 11 億個の予測構造を含む巨大なアトラスがオープンソース化され、研究者や開発者がウェブ上で探索・利用できるようになった。
専門モデルへの挑戦と「苦い教訓」
タンパク質折りたたみは言語モデリングとは異なる問題であるという通説に対し、大規模データと計算リソースで訓練された汎用モデルが特殊なドメイン特化型モデル(AlphaFold3 など)を凌駕する可能性を示し、AI 分野の「苦い教訓」が生物学にも適用されることを示唆した。
MSA依存からの脱却とスケーリング仮説の採用
AlphaFold2 が MSA(多重アラインメント)に依存するのに対し、ESM は大規模な非教師あり学習でタンパク質間の関係を学び、抗体など MSA データが存在しない領域でも汎化性能を発揮します。
タンパク質のための「ワールドモデル」の実現
ESM は現実世界のルールに従う抽象的なパターンを学習する「ワールドモデル」として機能し、これをベースに構造予測や機能設計などの専用ヘッド(Head)を接続することで多様なタスクに対応します。
生成と機械的解釈による新発見
このアプローチはタンパク質シーケンスの生成と実験室での検証、あるいは SAE などの手法を用いたモデル内部からの意味的特徴抽出を通じて、未知の生物学的知見を発見する道を開きます。
重要な引用
"The Bitter Lesson is Coming for Proteins"
"vanilla BERT-like transformer models trained on sufficiently large and diverse data sets can beat specialized models like AlphaFold3"
"World Model" is a hype term that I define like this: Use unsupervised training to learn abstract patterns from the data.
ESM takes a different approach: learn the relationship between different proteins by unsupervised training on as much diversity as you can find... and then correlate that back to structures known from the Protein Data Bank.
Like functions, the SAE features have a hierarchical composition from local, secondary and tertiary structures... but also motifs that are conceptual, such as membrane integrations, disordered regions and disulfide bonds.
The model represents disorderedness itself as a concept, with sub-features for different IDR flavors (polyampholyte, polar tract, prion-like domain).
影響分析・編集コメントを表示
影響分析
このニュースは、バイオテクノロジー分野における AI のパラダイムシフトを告げる画期的な出来事です。AlphaFold 系列のような高度に専門化されたアプローチに対する「計算リソースと大規模データによる汎用モデルの優位性」という、AI 業界で長年議論されてきた仮説が生物学でも実証されました。これにより、研究コストの削減や新薬開発の加速が期待され、タンパク質設計の民主化とスピードアップに大きく寄与するでしょう。
編集コメント
「苦い教訓」の概念が生物学に適用されるという衝撃的な発表であり、専門特化型モデルへの依存から、計算リソースとデータ規模を重視するアプローチへパラダイムがシフトしつつあることを示しています。
編集者注:Priscilla と Mark を迎えた最初の BioHub ポッドキャストにおいて、彼らは Alex Rives が率いる EvoScale の買収について議論しました。Alex は現在、BioHub の科学責任者を務めています。ESM-1 では、生命全体から収集した数百万のタンパク質配列を言語モデルとして訓練し、「次のトークン予測」というシンプルな目的で、ランダムにマスクされたアミノ酸を、残りの配列の文脈に基づいて予測しました。しかし彼らはすぐに、これらのモデルが生物学的構造や機能も学習することを発見しました。これはモデルが明示的に教えられていない性質も含み、この能力は計算資源に応じて予測可能にスケールすることです。これが ESM2 や ESM3 の開発へとつながりました。
本日、Alex はタンパク質生物学における予測、設計、発見を推進するためのオープンな科学エンジン「ESMFold 2」を発表しました。
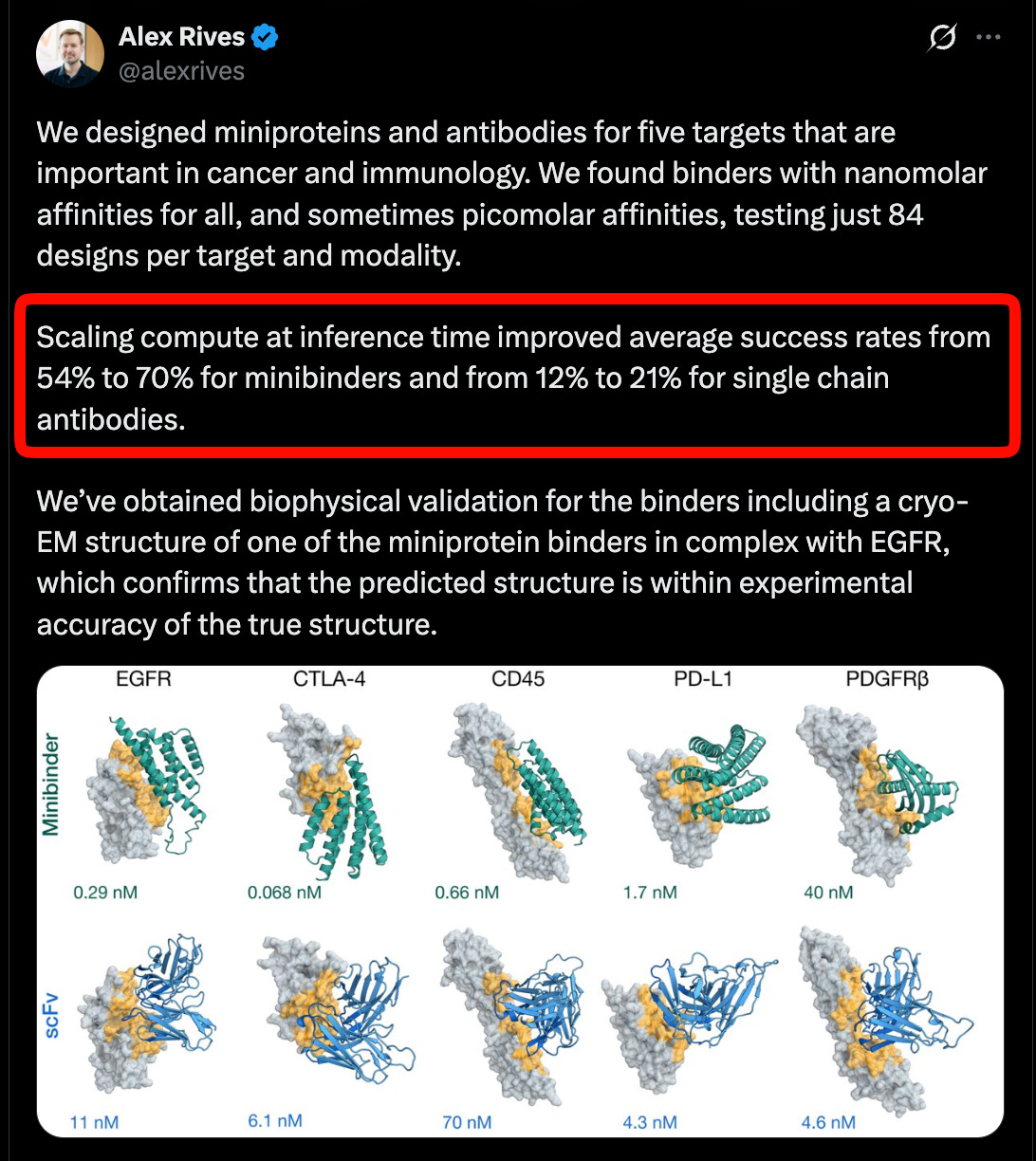
CZI ポッドキャストで議論されたクライオ電子顕微鏡(Cryo-EM)データに基づき、ESMFold2 は特に抗体という治療薬にとって重要なモダリティにおいて、タンパク質相互作用に関する最先端の性能を示し、がんおよび免疫学における 5 つの標的でも推論時間のスケール効果が機能していることを示す証拠を提供しました。
あの有名な AI×タンパク質折りたたみプロジェクトへの敬意を表して、彼らはまた 68 億個のタンパク質と 11 億個の予測構造からなるアトラスも公開しており、そのウェブサイト上で自由に探索することができます。この大規模なリリースに携わらせていただけたことを光栄に思います!
Science ポッドキャストで繰り返し耳にしてきた主張の一つに、「タンパク質折りたたみ、材料設計、細胞生物学などは、言語モデリングとは全く異なる問題である」というものがあります。確かにその通りです。しかし、BioHub の Alex Rives と ESM チームが最近発表したプレプリントとモデルは、十分に大規模で多様なデータセットで訓練されたバニラの BERT 型トランスフォーマーモデルが、AlphaFold3 などの専門モデルを上回る性能を、最も困難なタンパク質関連問題のいくつかにおいて示すことを実証しました。
Andrew White は、私たちの最初の LS-Science エピソードで素晴らしいセグメントを担当し、2020 年に AlphaFold2 が発表された際の驚異的な成果について解説しました。それは、DESRes がカスタム ASIC(特定用途向け集積回路)スーパーコンピュータクラスターを構築して解決しようとしていた問題を、デスクトップの GPU で突然解決可能にしたのです。この功績により、John Jumper と Demis Hassabis は化学賞ノーベル賞を受賞しました。
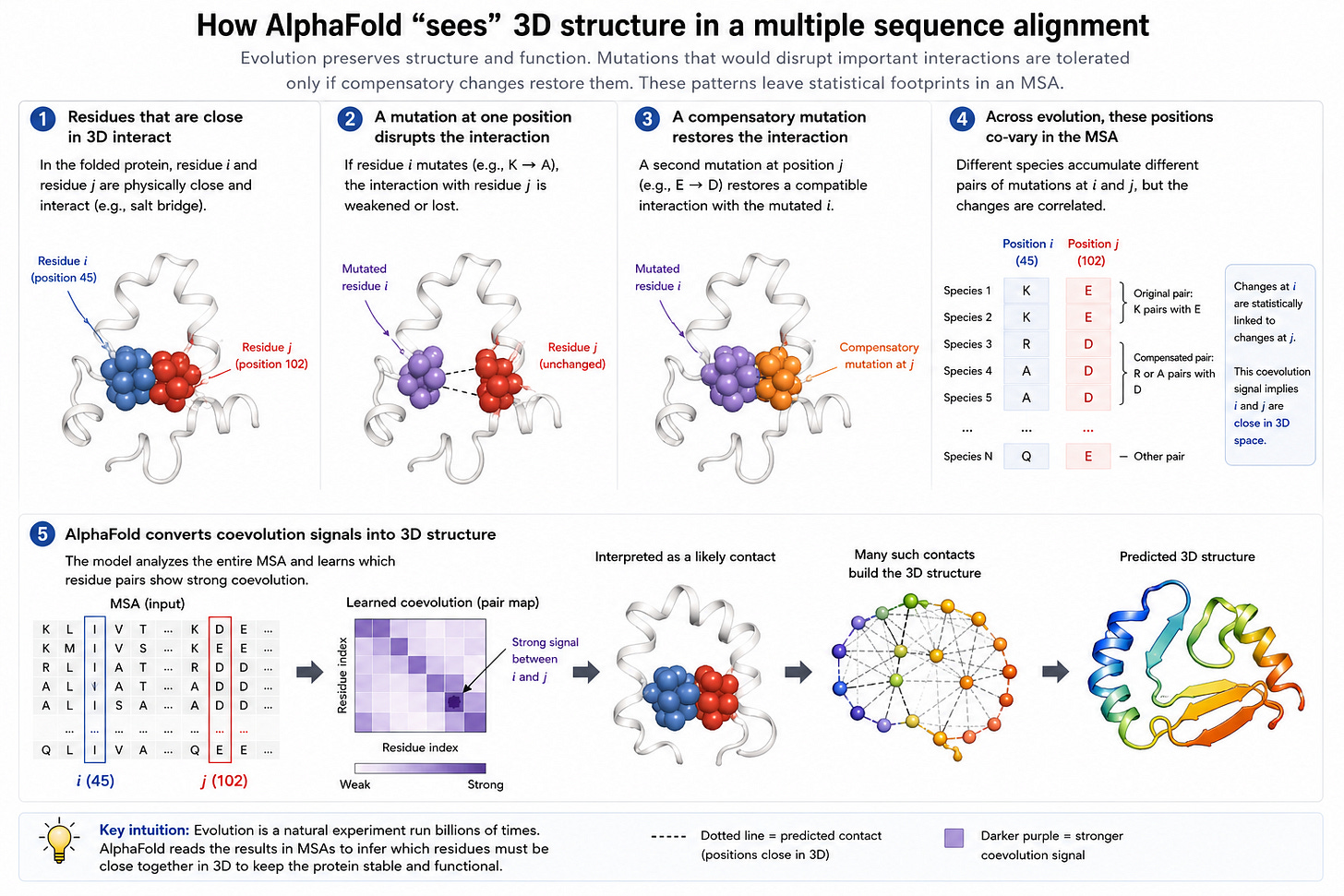
AlphaFold2 は非常に賢明な観察を利用しました:複数の種において突然変異のペアが共進化している場合、これはその突然変異が 3 次元空間上で近接するタンパク質の部分に対応することを意味します。これは通常、MSA(マルチシーケンスアライメント)と略され、AlphaFold2 をこれほど効果的なものにする鍵となる洞察です。
しかし、他の帰納的バイアスと同様に、これは一般化能力を損なうことになります。
流行になる前にスケールに傾倒した
LLM のスケーリング則のタイムラインと構造予測モデルのリリース(注 1)を見れば、ESM チームは AlphaFold2 が発表された後、MSA を無視するアプローチにさらに賭けたことがわかります。これは明らかにスケール仮説に対する多大な信念を必要とするものです。
なぜその確信があったのか?
ESM は、多くのスケーリング則と「苦い教訓(Bitter Lesson)」がますます正しく証明されていた時期に開発されました。AlphaFold2 の驚異的な成功は、同時に興奮させるとともに、苦々しい失望をもたらしたはずです。しかし、MSA を使用することは、モデルが特定のドメインで正確であるためには MSA を含むトレーニングデータに依存することを意味します。MSA を学習対象として持たない抗体のようなものについては、AlphaFold は概してうまく機能しません(注 2)。
ESM は異なるアプローチを採用しています:見つかる限り多様なデータを用いて教師なし学習を行い、タンパク質間の関係を学び(お馴染みの話ですね?)、その後、タンパク質データバンク(PDB)や他のソースから既知の構造と相関付けます。
つまり、これは「ワールドモデル」です。
タンパク質のためのワールドモデル
「ワールドモデル」という用語は私が以下のように定義する流行語です:
教師なし学習を用いてデータから抽象的なパターンを学習する:
- その抽象化は意味的であるべきで、新規の構築物は現実世界の法則に従うものを表す
- その抽象化は構成性を持つべきで、異なるパターンの再結合により、新規かつしばしば有効な構築物が導かれる
- その抽象化は一般化をサポートすべきで、学習していない現実世界のものも予測できる
一度ワールドモデルが完成すれば、下流タスクのために「ヘッド」を接続できます:タンパク質の性質を予測する、機能的特徴を分解する、あるいは設計基準を満たすタンパク質を検索するために表現を探る。BioHub が MIT ライセンスの下で直近リリースした 2 つの大規模モデルは、まさにこれに直接対応しています:
ワールドモデル → ESMC(28 億配列で訓練されたモデル)
構造予測ヘッド → ESMFold2
このワールドモデルが「ものを予測する」興味深い方法の一つとして、タンパク質配列を生成し、その後、実験室において結合親和性などの予測された性質を測定することが挙げられます。エピソードの中で Alex は、湿式実験(ウェットラボ)で検証した、彼らが予測した難易度の高い分子の一部について語っています。非常に素晴らしいことです!
もう一つの方法は、Sparse Auto Encoders (SAEs) などのメカニズム解釈(mech-interp)技術を用いてモデルから意味特徴を抽出し、未知の生物学を予測する新たな特徴を見つけることです。この部分についてはここではネタバレしません:私にとってこれはエピソードのハイライトの一つでした!
細胞はコンピュータである
私たちは皆、「遺伝子はコンピュータプログラムのようなものだ」という話を聞いたことがあるでしょうが、通常その比喩はそこで終わってしまいます。もちろん遺伝子は RNA に転写され、RNA はタンパク質に翻訳されるので、遺伝子はタンパク質を構築するためのプログラムですが、それは比喩を「2 進数がプログラムである」レベルまでしか持ち上げません。
より良い比喩があります:細胞核をストレージデバイス/ストレージコントローラーとみなし、リボソームを JIT コンパイラおよびランタイムとみなし、SAEs を通じて私たちの世界モデルから学習する意味特徴を関数とみなし、タンパク質をワークフロー(シグナル伝達経路)内で互いに相互作用して振る舞いや出力(表現型)を生み出すプロセスとみなすことができます。
関数と同様に、SAE 特徴は局所的、二次的、三次構造(タンパク質構造の模倣)4 から階層的に構成されていますが、膜統合、無秩序領域、ジスルフィド結合5 など概念的なモチーフも含まれます。これらの特徴を組み合わせて新たなタンパク質設計を行うにつれて、私たちはプログラム可能な生物学へとさらに近づいていきます。
アレックスはこの点についてエピソードの中でより詳細に説明しています。また以下のトピックについても触れています:
新しいデータ収集の原則
BioHub のビジョン
細胞のモデリング
お楽しみください!
フル動画ポッドキャスト
ご視聴いただきありがとうございます!
X: https://x.com/alexrives
LinkedIn:
1 image
image
抗体は、表面に新規タンパク質を有する病原体に適応するために非常に急速に変異します。この動的な性質により、多配列アライメント(MSAs: Multiple Sequence Alignments)は抗体には現れません。
これには、ESMC 用に AlphaFold2 自体を使用して作成されたデータセットも含まれており、これは AlphaFold の蒸留(distillation)であり、間接的には MSAs 自体にも依存しています。
非常に局所的な特徴(1〜3 アミノ酸残基): 個々のアミノ酸の生化学的特性、疎水性対極性の特徴、電荷
短距離の特徴(約 5〜10 アミノ酸残基): 二次構造 — αヘリックスの特徴、βストランドの特徴、βターンの特徴
中距離の特徴(約 10〜30 アミノ酸残基): 超二次構造モチーフ — βヘアピン、ヘリックスターンヘリックス、β-α-βユニット
長距離の特徴(タンパク質全体): 完全なドメイン識別子 — イムノグロブリンフォールド、ロスマンフォールド、TIM バレル、4 ヘリックスバンドル
DNA 結合特徴 — ヘリックスターンヘリックスタンパク質、ジンクフィンガー、ルイシンジッパー、および機能は共有するが配列は共有しない他の DNA 結合フォールドで活性化されます。
膜統合特徴 — GPCR、トランスポーター、またはチャネルのいずれに位置するかに関わらず、トランス膜セグメント上で活性化されます。
不規則領域の特徴:SAE は、予測する構造を持たない内在性不規則領域(IDR)に対して、約 686 の特徴(特徴予算の 5〜10%)を割当てています。これは驚くべき点です。モデルは「不規則性」そのものを概念として表現し、異なる IDR の種類(ポリアンフォライト、極性トラクト、プリオン様ドメインなど)に対応するサブ特徴も備えています。
ジスルフィド結合の特徴:ジスルフィド結合を形成するシステイン残基で活性化され、遊離のシステイン残基と区別されます。
原文を表示
Editor’s note: In our first BioHub pod with Priscilla and Mark they discussed their acquisition of EvoScale, led by Alex Rives, who is now Head of Science at BioHub. With ESM-1 they trained language models on millions of protein sequences drawn from across life, with a simple “next token” objective: predict the amino acids that have been randomly masked out, based on the context of the rest of the sequence. But they soon found that these models also learned biological structure and function, including properties the model had never been explicitly shown AND that this ability scales predictably with compute, leading to ESM2 and ESM3.
Today, Alex announced ESMFold 2, an open scientific engine to power prediction, design, and discovery across protein biology.
Building on Cryo-EM data (discussed in the CZI pod), ESMFold2 reports state of the art performance on protein interactions, especially antibodies, a critical modality for therapeutics, and evidence that inference time scaling is also working across five targets in cancer and immunology.
In a nod to that other famous AI x protein folding project, they are also releasing an atlas of 6.8 billion proteins, and 1.1 billion predicted structures, which you can play around with on their website. We are honored to work with them for this huge release!
One of the refrains we’ve heard on the Science pod has been that protein folding, materials design, cellular biology, etc. are very different problems from Language Modeling. They definitely are. Yet Alex Rives and the ESM team at BioHub just released a preprint and model, demonstrating that vanilla BERT-like transformer models trained on sufficiently large and diverse data sets can beat specialized models like AlphaFold3 on some of the hardest protein-related problems.
Andrew White had a great segment in our first LS-Science episode that explained how mind blowing AlphaFold2 was when it was released in 2020: it suddenly solved problems on a GPU on your desktop that DESRes had built custom-ASIC supercomputer clusters to solve. John Jumper and Demmis Hassabis received the Nobel Prize in Chemistry for this work.
AlphaFold2 took advantage of an very clever observation: if multiple species co-evolve pairs of mutations, this implies that the mutations correspond to parts of the protein that are close in 3d space. This is usually shorthanded as MSAs (multi-sequence alignments), and is the key insight which makes AlphaFold2 so effective.
Like other inductive biases, however, it hurts generalization.
Scale-pilled before it was cool
If you take a look at the timeline for scaling laws for LLMs and release of structure prediction models1, the ESM team notably doubled down on their MSAs-be-damned approach after AlphaFold2 released. This obviously requires a great deal of belief in the scale hypothesis.
Why the conviction?
ESM developed at a time when many of the scaling laws and the “Bitter Lesson” were proving increasingly correct. AlphaFold2’s wild success must have been both exciting and bitterly disappointing. But using MSAs mean that the model is is dependent on training data that contains MSAs in order to be accurate in a given domain. For things like antibodies that don’t have MSAs to train on2, AlphaFold tends to do poorly.
ESM takes a different approach: learn the relationship between different proteins by unsupervised training on as much diversity as you can find (sound familiar?) and then correlate that back to structures know from the Protein Data Bank (PDB) and other sources3.
In other words, a World Model.
World Model for proteins
“World Model” is a hype term that I define like this:
Use unsupervised training to learn abstract patterns from the data:
The abstraction should be semantic - novel constructions represent things that obey the rules of the real world
The abstraction should be compositional - recombining different patterns leads to novel and often valid constructions
The abstraction should support generalization - it predicts things in the real world it wasn’t trained on
Once you have a world model, you can attach “heads” to it for downstream tasks: predict properties of a protein, decompose its functional features, or search the representation for proteins that meet design criteria. The two big models BioHub just released under MIT license map directly onto this:
World model → ESMC (a model trained on 2.8 billion sequences)
Structure-prediction head → ESMFold2
One of the interesting ways the world model can “predict things” is to generate proteins sequences and then measure the predicted properties, such as binding affinity, in the lab. Alex talks in the episode about validating some of the harder molecules they predicted in the wet-lab. Very cool!
Another way is to use mech-interp techniques such as Sparse Auto Encoders (SAEs) to extract semantic features from your model, and then find novel features that predict unknown biology. I won’t spoil this part for you: it was one of the highlights of the episode for me!
A cell is a computer
We have all heard that genes are like computer programs, but usually the analogy fizzles after that. Of course genes are transcribed into RNA and RNA is translated into proteins, so genes are programs for building proteins, but that carries the analogy only to “binary digits are programs.”
Here’s a better analogy: you can think of the cell nucleus as a storage device / storage controller, the ribosome as a JIT-compiler and runtime, and the semantic features that we learn from our world model via SAEs as functions, proteins as processes that interact together in workflows (signalling pathways) to produce behaviors and outputs (phenotypes).
Like functions, the SAE features have a hierarchical composition from local, secondary and tertiary structures (mimicing protein structure)4, but also motifs that are conceptual, such as membrane integrations, disordered regions and disulfide bonds5. As we learn to compose these features we into novel protein designs, we move further towards programmable biology.
Alex goes into much more detail about this in the episode, as well as:
Principles for new data collection
BioHub’s vision
Modeling the cell
Enjoy!
Full Video podcast
please like and subscribe!
X: https://x.com/alexrives
LinkedIn:
1image
2Antibodies mutate very rapidly so that they can adapt to pathogens with novel proteins on them. These dynamics mean that MSAs don’t appear in them.
3This includes a dataset created using AlphaFold2 itself for ESMC, making it a distillation of AlphaFold, and indirectly dependent on MSAs itself.
4Very local (1–3 residues): individual amino acid biochemistry, hydrophobic vs. polar character, charge
Short-range (~5–10 residues): secondary structure — α-helix features, β-strand features, β-turn features
Medium-range (~10–30 residues): supersecondary motifs — β-hairpins, helix-turn-helix, β-α-β units
Long-range (whole-protein): full domain identifiers — immunoglobulin fold, Rossmann fold, TIM barrel, four-helix bundle
5DNA-binding features — activated across helix-turn-helix proteins, zinc fingers, leucine zippers, and other DNA-binding folds that share function but not sequence
Membrane integration features — activated on transmembrane segments regardless of whether they sit in a GPCR, a transporter, or a channel
Disordered region features the SAE devotes ~686 features (5–10% of the feature budget) to intrinsically disordered regions, which is striking because IDRs have no structure to predict. The model represents disorderedness itself as a concept, with sub-features for different IDR flavors (polyampholyte, polar tract, prion-like domain)
Disulfide bond features — activated on cysteines that participate in disulfides, distinguishing them from free cysteines
関連記事
今日のまとめ
AI日報で今日の重要ニュースをまとめ読み