MatterSim を用いた材料開発の進展:実験的合成と高速シミュレーション、マルチタスクモデル
Microsoft Research は、MatterSim の予測を実験で検証し、シミュレーション速度を大幅に向上させるとともに、多機能な新モデル「MatterSim-MT」を発表することで、材料設計プロセスの加速と精度向上を実現した。
キーポイント
実験的検証による予測精度の実証
MatterSim-v1 の計算予測に基づき合成された四角錐形リン化タンタル(TaP)の熱伝導率が、シミュレーション結果とほぼ一致する 152 W/m/K であることを実験で確認した。
シミュレーション速度の劇的向上
MatterSim-v1 の推論を 3〜5 倍高速化し、LAMMPS ソフトウェアパッケージと統合することで、複数の GPU を活用した大規模シミュレーションが可能になった。
多機能基盤モデル「MatterSim-MT」の登場
単純なポテンシャルエネルギー表面を超え、複雑な多物性現象をシミュレートできる新しいマルチタスク基盤モデルを発表し、材料特性評価の幅を広げた。
大規模材料スクリーニングの実現
MatterSim-v1を用いて24万を超える候補材料を熱伝導率の高い物質としてスクリーニングし、従来の手法では不可能だったスケールでの探索を可能にしました。
高精度かつ高効率な予測
第一原理計算と高い一致を示す精度を持ちながら計算コストが低いため、熱伝導率のような感度の高い性質を大規模に評価する鍵となりました。
実験的検証による熱伝導体発見
MatterSim の予測に基づきテトラゴン型リン化タンタル (TaP) を合成・評価した結果、シリコンに匹敵する高い熱伝導率(152 W/m/K)を確認し、機能性材料の探索可能性を実証しました。
推論速度の大幅向上と LAMMPS 統合
グラフ構築の高速化や事前コンパイルなどの最適化によりモデル推論が最大 5 倍速くなり、LAMMPS シミュレーションソフトウェアへの統合でマルチ GPU スケーリングが可能になりました。
影響分析・編集コメントを表示
影響分析
この発表は、AI を活用した材料設計が「予測段階」から「実験検証済み・実用化直前」のフェーズへ移行したことを示す重要な転換点です。特にシミュレーション速度の向上と多機能モデルの導入により、従来の第一原理計算では不可能だった大規模かつ複雑な材料探索が短時間で可能になり、ナノエレクトロニクスやエネルギー貯蔵分野での技術革新を加速させる可能性があります。
編集コメント
AI が単なる予測ツールではなく、実験結果と連動して材料開発のサイクルを劇的に短縮する実証段階に入ったことを示す画期的なニュースです。特に速度向上と多機能化は、産業応用への道筋を明確にしました。

一言で言うと
実験的検証:MatterSim-v1 を用いたハイスループットスクリーニングにより、以前は四角錐型のリン化タンタル(TaP)が高性能熱伝導体として有望であることを特定しました。今回、実際にこれを合成し、その熱伝導率を測定したところ、シリコンの熱伝導率に近い値(152 W/m/K)であることが確認されました。
シミュレーションの高速化:MatterSim-v1 モデルの推論速度を 3〜5 倍に加速し、LAMMPS ソフトウェアパッケージと統合しました。これにより、複数の GPU を跨ぐ大規模シミュレーションが可能になりました。
新モデルの公開:in silico(コンピュータ上での)材料特性評価のためのマルチタスク基盤モデル「MatterSim-MT」を発表します。このモデルは、ポテンシャルエネルギー面だけでは捉えきれない複雑な多物性現象のシミュレーションを可能にします。
材料設計は、ナノエレクトロニクスから半導体設計、エネルギー貯蔵に至るまで、幅広い技術的進歩の基盤となっています。しかし、新規材料の開発サイクルはまだ遅く、コストも高いままです。汎用機械学習原子間ポテンシャル(interatomic potentials)は、広範な材料に対する正確な安定性と物性の予測を提供することで、材料設計プロセスを加速することを目指しています。これらのモデルは、従来の第一原理シミュレーション(first-principles simulations)に比べて桁違いに高速であり、以前は非現実的だった問題を数時間で完了できる日常的な計算へと変えています。MatterSim-v1 モデルの公開以来、有限温度や圧力といった現実的な条件下での材料を正確にシミュレートする能力により、材料科学コミュニティで人気を集めています。
本日、私たちはいくつかの刺激的な MatterSim のアップデートを発表します。これには、熱伝導体に対する MatterSim 予測の実験的検証、より高速なシミュレーションのためのパフォーマンス向上、および材料特性評価のための新しいマルチタスク基盤モデル(multi-task foundation model)の導入が含まれます。
実験的検証
 image図 1:MatterSim の計算予測に基づき、潜在的な高熱伝導体を合成しました。左側:熱伝導率に関する MatterSim の予測値と、基準となるシミュレーションおよび実験結果の比較(参考のために±50% の誤差範囲を示しています)。右側:測定された熱伝導率が 152 W/m/K である実験的に合成された正方晶タングステンリン化物 (TaP) サンプルの異なる視点からの画像。
image図 1:MatterSim の計算予測に基づき、潜在的な高熱伝導体を合成しました。左側:熱伝導率に関する MatterSim の予測値と、基準となるシミュレーションおよび実験結果の比較(参考のために±50% の誤差範囲を示しています)。右側:測定された熱伝導率が 152 W/m/K である実験的に合成された正方晶タングステンリン化物 (TaP) サンプルの異なる視点からの画像。
高い熱伝導率を持つ材料は、過熱防止やエネルギー効率の向上といった熱管理において重要な役割を果たします。例えば、ダイヤモンド、銅、シリコンといった確立された高熱伝導体は、幅広い冷却応用分野で広く利用されています。次世代の高熱伝導体を設計することは、コンピューティング、パワーエレクトロニクス、航空宇宙技術における進展を可能にするかもしれません。しかし、それを実現するには、候補材料の熱伝導率値を正確に予測する必要があります。
固体中では、熱は主に 2 つの方法で運ばれます:振動する原子(フォノン)によるものと、移動する電子によるものです。フォノンの寄与は、機械学習を用いた原子間ポテンシャルによって推定することができ、これにより数千もの候補をスクリーニングして、高価な実験的検証を行う前に最も有望な材料に探索範囲を絞り込むことが可能になります。
「MatterSim は、これまでに計算された熱伝導率のデータベースの中で最も大規模なものを生成しました。これは、以前よりもはるかに広い材料空間を探求する扉を開くものです。」
– テキサス大学ダラス校 ビン・リュウ教授
テキサス大学ダラス校(UT Dallas)、イリノイ大学アーバナ・シャンペーン校、カリフォルニア大学デービス校(UC Davis)との共同研究において、MatterSim-v1 を用いて高熱伝導体となる 240,000 以上の候補材料をスクリーニングしました。図 1(左側)に示す通り、MatterSim の予測は第一原理シミュレーション [first-principles simulations] と良好な一致を示しています。UC Davis のダヴィデ・ドナディオ教授は次のように述べています。「MatterSim モデルが、熱伝導率のような感度の高い性質を予測する際に、いかに精度と計算効率を両立させたかには驚きました。これが、従来の方法では全く手が届かなかった規模、すなわち数十万もの結晶体をスクリーニングすることを可能にした鍵でした。」UT Dallas のビン・リュウ教授はさらに、「MatterSim はこれまでにないほど大規模な計算熱伝導率データベースを生成しました。これは、以前よりもはるかに広い材料空間を探求する扉を開くものであり、実用的な要件を満たすことを課した後も、コミュニティがより広範な実現可能な材料群を発見することを可能にします。」と述べています。
「初めて、熱伝導率を支配する要因に関する従来の理解を大規模に検証することができるようになりました。」
– イリノイ大学アーバナ・シャンペーン校 デイビッド・カヒル教授
これらの予測に基づき、四角晶のリン化タンタル(TaP)が高熱伝導体としての可能性を有する材料であることを特定しました。私たちは UT Dallas で四角晶のリン化タンタル(TaP)を実験的に合成し、イリノイ大学アーバナ・シャンペーン校でその熱伝導率を測定しました(最良の試料で 152 W/m/K)。これはシリコンの熱伝導率に近い値です。四角晶の TaP の合成自体は私たちが初めてではありませんが、この材料が熱伝導体として検討されたことはこれまでありませんでした。これらの結果は、MatterSim が機能性材料の特定をどのように可能にするかを示しています。「初めて、熱伝導率を支配する要因に関する従来の理解を大規模に検証できるようになり、質量密度、元素の豊富さ、環境安定性といった他の重要な制約とのバランスを保ちながら新たな機能性材料を発見することが可能になります」と、イリノイ大学アーバナ・シャンペーン校のデビッド・カヒル教授は述べています。
PODCAST SERIES

医療における AI 革命、再考
マイクロソフトのピーター・リーと共に、AI がヘルスケアにどのような影響を与え、医学の未来にとって何を意味するのかを探る旅に出ましょう。
今すぐ聴く
新しいタブで開きます
パフォーマンスの向上
MatterSim-v1 のパフォーマンスと使いやすさを向上させるためのオープンソースの改善策を複数公開することで、MatterSim-v1 を大幅に高速化しています。まず、グラフ構築の高速化、事前コンパイルの実装、原子表現間の変換削減を組み合わせてモデル推論を加速し、MatterSim-v1.0.0-5M では 3 倍、MatterSim-v1.0.0-1M では 5 倍の速度向上を実現しました(図 2 を参照)。MatterSim-v1 の利用をより容易にするため、広く使用されている LAMMPS シミュレーションソフトウェアに統合し、既存のワークフロー内で複数の GPU にわたってモデル推論を簡単にスケーリングできるようにしました。
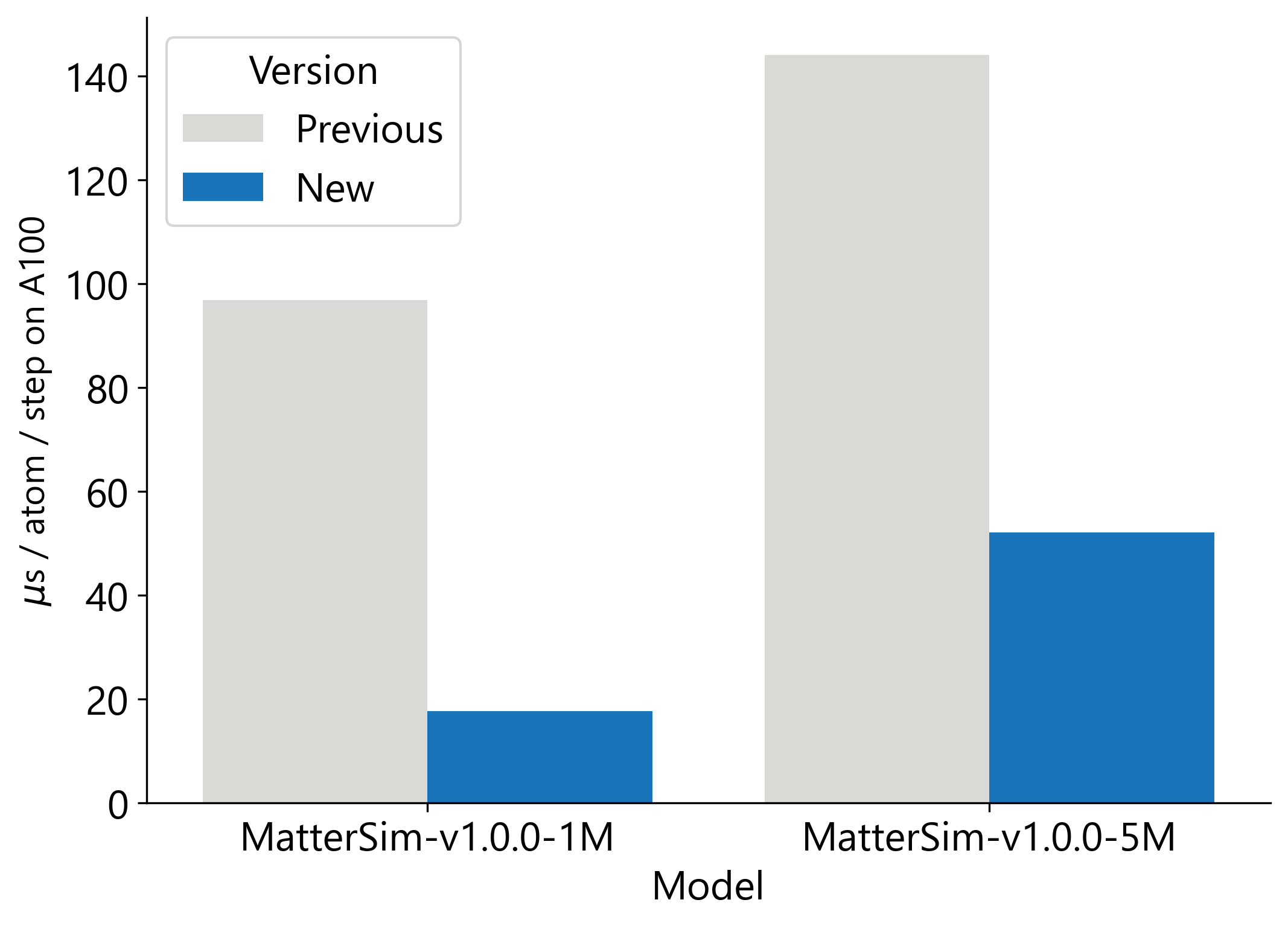 image図 2: MatterSim-v1.0.0-5M の推論速度が 3 倍、MatterSim-v1.0.0-1M の推論速度が 5 倍に向上(Python)。
image図 2: MatterSim-v1.0.0-5M の推論速度が 3 倍、MatterSim-v1.0.0-1M の推論速度が 5 倍に向上(Python)。
新モデルのリリース
MatterSim-v1 の成功を踏まえ、本日は MatterSim モデルファミリーを拡張し、in silico 材料シミュレーションおよび物性評価のためのマルチタスク (MT) ファウンデーションモデルである MatterSim-MT を発表します。このモデルは、エネルギー、力、応力、および重要な材料特性をネイティブに予測できます。
MatterSim-MT は、89 元素にわたる 3500 万構造以上を第一原理ラベル付きデータとして事前学習しており、温度は最大 5000 K、圧力は最大 1000 GPa に至ります。さらに、バダー電荷(Bader charges)、磁気モーメント、ボーン有効電荷、誘電行列など、さまざまな物性値を用いて微調整が施されています。箱から出したままの状態でも、MatterSim-MT は材料の構造・ダイナミクス・熱力学を予測するためのファウンデーションモデルとして機能します。そのマルチタスクアーキテクチャは、ポテンシャルエネルギー面だけでは捉えきれない多様な複雑なシミュレーションも可能にします。これらの現象を正確にシミュレートする能力は、触媒やエネルギー貯蔵などの応用において極めて重要です。
ここでは、振動分光法、強誘電体スイッチング、電気化学的酸化還元反応の 3 つの事例研究を通じて、これらのマルチタスク機能を具体化します。各例では、異なる組み合わせの物性予測が必要です。完全な論文稿においては、MatterSim-MT がより多くのデータとパラメータに対してよくスケーリングすること、理論レベルを高めるために効率的に微調整可能であること、アクティブラーニングを通じて体系的に新たな系へ拡張できることも示しています。
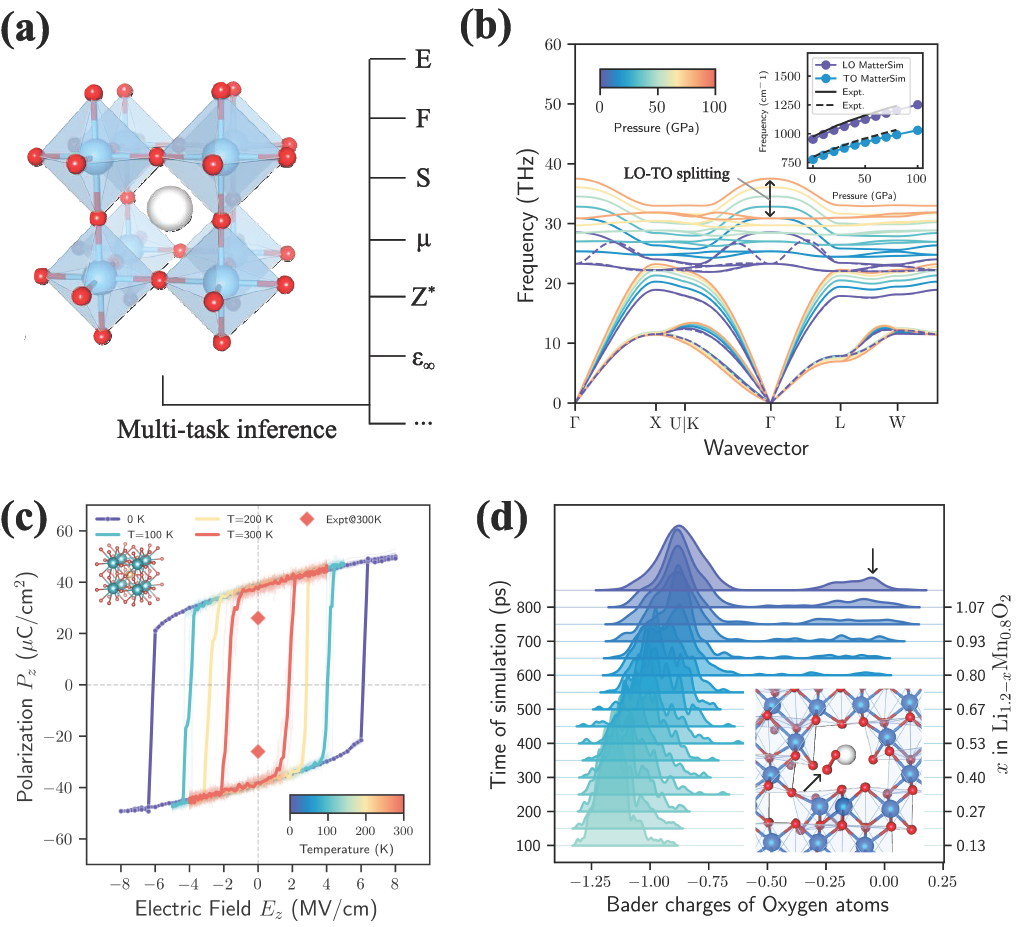 image図 3:MatterSim-MT の多タスク予測能力により、複雑な物質現象のシミュレーションが可能になります。(a) MatterSim-MT の多タスク推論機能の模式図。原子構造からエネルギー (E)、力 (F)、応力 (S)、磁気モーメント (μ)、Born 有効電荷 (Z∗)、および誘電行列 (ε∞) を予測する能力を含みます。(b) 100 GPa までの圧力依存性を持つ炭化ケイ素 (SiC) のフォノンスペクトル。挿入図では、MatterSim が予測した縦光学 (LO) および横光学 (TO) モードの分裂と実験測定値を比較しています。(c) 強誘電性正方晶 BaTiO3 材料において、z 方向の電気場に対する分極密度のヒステリシス曲線の予測結果。(d) リチウム脱離過程における Li1.2 – xMn0.8O2 の酸素 Bader 電荷分布の変化。矢印は O2 分子の生成を示しています。
image図 3:MatterSim-MT の多タスク予測能力により、複雑な物質現象のシミュレーションが可能になります。(a) MatterSim-MT の多タスク推論機能の模式図。原子構造からエネルギー (E)、力 (F)、応力 (S)、磁気モーメント (μ)、Born 有効電荷 (Z∗)、および誘電行列 (ε∞) を予測する能力を含みます。(b) 100 GPa までの圧力依存性を持つ炭化ケイ素 (SiC) のフォノンスペクトル。挿入図では、MatterSim が予測した縦光学 (LO) および横光学 (TO) モードの分裂と実験測定値を比較しています。(c) 強誘電性正方晶 BaTiO3 材料において、z 方向の電気場に対する分極密度のヒステリシス曲線の予測結果。(d) リチウム脱離過程における Li1.2 – xMn0.8O2 の酸素 Bader 電荷分布の変化。矢印は O2 分子の生成を示しています。
まず、振動分光法に焦点を当てます。これは原子結合の自然な振動を測定することで物質を同定する技術です。我々は、Born 有効電荷と誘電特性の予測が、極性結晶におけるフォノンスペクトルの計算を可能にする方法を実証します。これらの材料では、反対の電荷を持つイオンがお互いに振動し合います。振動の方向によっては、電荷が蓄積して巨視的な電気場が生じ、光学フォノンモードが高周波数の縦波 (LO) と低周波数の横波 (TO) の枝に分裂します。事例研究として、高電力電子機器に使用される材料である 3c-炭化ケイ素 (3c-SiC) において、極端な圧力下でのこの挙動をシミュレーションしました。Fig. 3(b) に示す通り、MatterSim-MT は理論値および実験値と非常に良く一致する Born 有効電荷を予測しています。その結果生じる LO-TO 分裂は 5.26 THz で、第一原理計算からは 0.06 THz のみ、実験測定からは 0.03 THz のみ乖離しています。
予測されたボーン有効電荷を用いることで、系が外部電場に対してどのように応答するかをシミュレーションすることも可能です。強誘電体材料では、イオンは非対称な配列をとることで結晶に正味の電気分極を与え、これが印加電場によって反転します。図 3(c) では、印加電場下でのチタン酸バリウム(BaTiO3)をシミュレーションし、その分極のスイッチングを再現しました。得られたヒステリシス曲線は、300 K における有限温度効果により分極の反転が容易になることを正しく示しており、予測された自発分極(38 μC/cm2)が実験値(26 μC/cm2)よりわずかに高いにもかかわらずです。この不一致は、基礎となる第一原理計算でよく知られている結合不足に起因するものと考えられます。
最後に、化学結合や酸化還元反応における電子の自由度を研究するために原子電荷を予測します。私たちは、シミュレーションされたバッテリー充電プロセス中のカソード材料 Li1.2 – xMn0.8O2 の挙動を検討しています。これらのリチウム豊富な遷移金属酸化物は高いエネルギー密度を持つため次世代電池として有望ですが、陰イオン酸素酸化還元メカニズムに起因する不可逆的な容量損失という課題を抱えています。私たちは 1000 K で分子動力学シミュレーションを実行し、バッテリー充電を模倣するためにリチウムを段階的に抽出することで、この現象を再現しました。時間の経過とともに明確な変化を観察します:最初はマンガン(Mn)原子が充電に必要な電子を供給しますが、より多くのリチウムが除去されると、酸素原子が代わりに電子を放出することを余儀なくされます(陽イオンから陰イオンの酸化還元)。これは、時間とともに Bader 電荷が負の値から減少するシフトによって示されています(Fig. 3(d))。これにより構造が不安定化し、酸素原子が対になって O2 二量体を形成します(Fig. 3(d)、インセット)。注目すべきは、この陽イオンから陰イオンの酸化還元遷移と格子劣化の包括的な図景が、電池材料に対するタスク固有のトレーニングを一切行わずに、マルチタスク予測から自然に導き出される点です。
次のステップ
実験的検証、大幅な性能向上、そして新たなマルチタスク機能により、MatterSim は材料設計におけるより実用的で意思決定に直結する用途へと進化を遂げています。これらの進展は、材料科学者が大規模な計算スクリーニングから標的型の実験フォローアップおよび意思決定に関連する科学的ワークフローへ、より迅速に移行することを支援しています。材料科学コミュニティがこれらの進歩をそれぞれの領域でどのように応用するか、楽しみに見ています。
MatterSim がテストされ、拡張され、現実世界の材料発見パイプラインに統合される過程での継続的な協力を楽しみにしています。
MatterSim の更新
MatterSim-MT プリプリント(査読前論文)
謝辞
本稿は、Microsoft Research AI for Science が主導し、Microsoft Research Accelerator と協力し、テキサス大学ダラス校(MSR Accelerator の支援あり)、イリノイ大学アーバナ・シャンペーン校、カリフォルニア大学デービス校の共同研究者らとの連携により実現された、極めて協力的かつ学際的な取り組みの結果です。本稿への貢献者には、Han Yang, Xixian Liu, Chenxi Hu, Yichi Zhou, Yu Shi, Chang Liu, Junfu Tan, Jielan Li, Guanzhi Li, Qian Wang, Yu Zhu, Zekun Chen, Shuizhou Chen, Fabian Thiemann, Claudio Zeni, Matthew Horton, Robert Pinsler, Andrew Fowler, Daniel Zügner, Tian Xie, Lixin Sun, Yicheng Chen, Lingyu Kong, Yeqi Bai, Deniz Gunceler, Frank Noé, Hongxia Hao, Ziheng Lu, Zixin Zhai, Mengfan Wu, Haoke Qiu, Mingfa Tang, Tie-Yan Liu, Haiguang Liu, Tao Qin, David G. Cahill, Bing Lv, Davide Donadio, Shoko Ueda, Kenji Takeda が含まれます。
新しいタブで開きます。記事「MatterSim を用いた材料分野における AI の進展:実験的合成、高速シミュレーション、マルチタスクモデル」は、Microsoft Research で最初に発表されました。
原文を表示
At a glance
Experimental validation: Using high-throughput screening with MatterSim-v1, we previously identified tetragonal tantalum phosphorus (TaP) as a potential high-performance thermal conductor. Now we have experimentally synthesized it and measured its thermal conductivity (152 W/m/K) to be close to the thermal conductivity of silicon.
Faster simulation: We have accelerated MatterSim-v1 model inference by 3-5x and integrated it with the LAMMPS software package, enabling large-scale simulations across multiple GPUs.
New model release: We are introducing MatterSim-MT, a multi-task foundation model for in silico materials characterization that enables the simulation of complex, multi-property phenomena beyond what potential energy surfaces alone can capture.
Materials design underpins a wide range of technological advances, from nanoelectronics to semiconductor design and energy storage. Yet development cycles for novel materials remain slow and costly. Universal machine learning interatomic potentials aim to accelerate the materials design process by providing accurate stability and property predictions for a wide range of materials. These models are orders of magnitude faster than traditional first-principles simulations, turning previously impractical problems into routine computations that can be completed in a few hours. Since we launched our MatterSim-v1 model, it has gained popularity in the materials science community for its ability to accurately simulate materials under realistic conditions, including finite temperature and pressure.
Today, we have several exciting MatterSim updates to share. These include experimental validation of MatterSim predictions for thermal conductors, performance improvements for faster simulation, and the introduction of a new multi-task foundation model for materials characterization.
Experimental validation
imageFigure 1: Based on MatterSim’s computational predictions, we have synthesized a potential high thermal conductor. Left: MatterSim predictions of thermal conductivity compared to ground-truth simulation and experiment (with ±50% error band shown for reference). Right: Different views of the experimentally synthesized tetragonal tantalum phosphorus (TaP) sample with measured thermal conductivity of 152 W/m/K.
Materials with high thermal conductivity play a critical role in heat management, preventing overheating and improving energy efficiency. For example, established high thermal conductors like diamond, copper and silicon are widely used across a broad range of cooling applications. Designing next-generation thermal conductors may enable advances in computing, power electronics, and aerospace technologies. However, doing so requires accurate predictions of thermal conductivity values for candidate materials.
In solids, heat is carried in two main ways: by vibrating atoms (phonons) and by moving electrons. The phonon contribution can be estimated using machine-learning interatomic potentials to enable screening of thousands of candidates, narrowing the search space to the most promising materials before expensive experimental validation.
“MatterSim has generated by far the largest database of computational thermal conductivities. This opens the door to exploring a far broader materials space than before […].”
– Prof. Bing Lv, University of Texas Dallas
In collaboration with the University of Texas Dallas (UT Dallas), University of Illinois Urbana-Champaign, and University of California Davis (UC Davis), we have used MatterSim-v1 to screen over 240,000 candidate materials for high thermal conductors. As shown in Fig. 1 (left), MatterSim’s predictions have good agreement with first-principles simulations. Prof. Davide Donadio from UC Davis: “I was amazed by how the MatterSim model combined accuracy and computational efficiency to predict such a sensitive property as thermal conductivity. That was the key that unlocked screening at this scale, hundreds of thousands of crystals, that would have been completely out of reach with conventional methods.” Prof. Bing Lv from UT Dallas adds: “MatterSim has generated by far the largest database of computational thermal conductivities. This opens the door to exploring a far broader materials space than before, enabling the community to uncover a broader set of viable materials even after imposing practical requirements.”
“For the first time, we can test conventional understanding of what controls thermal conductivity at scale […]”
– Prof. David Cahill, University of Illinois Urbana-Champaign
Based on these predictions, we have identified tetragonal tantalum phosphorus (TaP) as a potential high thermal conductor. We have experimentally synthesized tetragonal tantalum phosphorus (TaP) at UT Dallas and measured its thermal conductivity at University of Illinois Urbana-Champaign (152 W/m/K for our best samples), close to the thermal conductivity of silicon. While we are not the first to synthesize tetragonal TaP, the material has not been considered as a thermal conductor before. These results demonstrate how MatterSim can enable the identification of functional materials: “For the first time, we can test conventional understanding of what controls thermal conductivity at scale, while enabling the discovery of new functional materials that balance it with other important constraints such as mass density, elemental abundance, and environmental stability”, says Prof. David Cahill from University of Illinois Urbana-Champaign.
PODCAST SERIES
image
The AI Revolution in Medicine, Revisited
Join Microsoft’s Peter Lee on a journey to discover how AI is impacting healthcare and what it means for the future of medicine.
Listen now
Opens in a new tab
Performance improvements
We are making MatterSim-v1 significantly faster by releasing several open-source performance and usability improvements. First, we speed up model inference through a combination of faster graph construction, ahead-of-time compilation and reduced conversion between atomic representations, resulting in a 3x speed-up of MatterSim-v1.0.0-5M and a 5x speed-up of MatterSim-v1.0.0-1M (see Fig. 2). To make MatterSim-v1 easier to use, we have integrated it into the widely used LAMMPS simulation software, allowing users to easily scale model inference across multiple GPUs in their existing workflows.
imageFigure 2: 3x inference speed-up of MatterSim-v1.0.0-5M and 5x inference speedup of MatterSim-v1.0.0-1M (python).
New model release
Building on the success of MatterSim-v1, today we extend the MatterSim model family by announcing MatterSim-MT: a multi-task (MT) foundation model for in silico materials simulation and property characterization. The model natively predicts energies, forces, stress and several important materials properties.
MatterSim-MT is pretrained on over 35 million first-principles-labelled structures covering 89 elements, temperatures up to 5000 K and pressures up to 1000 GPa. It is further fine-tuned on various properties including Bader charges, magnetic moments, Born effective charges, and dielectric matrices. Out of the box, MatterSim-MT serves as a foundation model for predicting material structure, dynamics and thermodynamics. Its multi-task architecture also enables a wide range of complex simulations that cannot be captured by potential energy surfaces alone. The ability to accurately simulate these phenomena is crucial for applications such as catalysis and energy storage.
Here, we illustrate these multi-task capabilities through three case studies: vibrational spectroscopy, ferroelectric switching, and electrochemical redox. Each example requires a distinct combination of property predictions. In the full manuscript, we also show that MatterSim-MT scales well with more data and parameters, can be efficiently fine-tuned to higher levels of theory, and can be systematically extended to new systems via active learning.
imageFigure 3: MatterSim‑MT’s multi-task prediction ability enables simulating complex material phenomena. (a) Illustration of the multi-task inference capabilities of MatterSim-MT, including predictions of energy (E), forces (F), stress (S), magnetic moments (μ), Born effective charges (Z∗), and dielectric matrices (ε∞) from atomic structures. (b) Pressure-dependent phonon spectrum of silicon carbide (SiC) up to 100 GPa, with inset comparing MatterSim’s predicted longitudinal optical (LO) and transverse optical (TO) splitting against experimental measurements. (c) Predicted hysteresis curve of polarization density as a function of the electrical field along the z direction in the ferroelectric tetragonal BaTiO3 material. (d) Evolution of oxygen Bader charge distributions in Li1.2 – xMn0.8O2 during delithiation, with arrows indicating the formation of an O2 molecule.
First, we focus on vibrational spectroscopy, a technique that identifies substances by measuring how their atomic bonds naturally vibrate. We demonstrate how predictions of Born effective charges and dielectric properties enable the computation of phonon spectra in polar crystals. In these materials, oppositely charged ions vibrate against each other. Depending on the direction of vibration, this can lead to a buildup of charge that creates a macroscopic electric field, splitting the optical phonon modes into higher-frequency longitudinal (LO) and lower-frequency transverse (TO) branches. As a case study, we simulated this behavior in 3c-silicon carbide (3c-SiC), a material used in high-power electronics, under extreme pressures. As shown in Fig. 3(b), MatterSim-MT predicts a Born effective charge in close agreement with both theoretical and experimental values. The resulting LO-TO splitting of 5.26 THz deviates by only 0.06 THz from ab initio calculations and 0.03 THz from experimental measurements.
The predicted Born effective charges also allow us to simulate how systems respond to an external electric field. In ferroelectric materials, ions adopt an asymmetric arrangement that gives the crystal a net electric polarization that can be flipped by an applied field. In Fig. 3(c), we demonstrate this by simulating barium titanate (BaTiO3) under an applied electric field, reproducing the switching of its polarization. The resulting hysteresis curve correctly shows that finite-temperature effects at 300 K make it easier to flip the polarization, even though the predicted spontaneous polarization (38 μC/cm2) is slightly higher than the experimental value (26 μC/cm2). This discrepancy is likely due to the well-known underbinding of the underlying first-principles calculations.
Finally, we predict atomic charges to study the electronic degrees of freedom in chemical bonding and redox processes. We examine the behavior of the cathode material Li1.2 – xMn0.8O2 during a simulated battery charging process. These lithium-rich transition-metal oxides are promising next-generation batteries due to their high energy density but suffer from irreversible capacity loss associated with the anionic oxygen redox mechanism. We reproduced this phenomenon by running molecular dynamics simulations at 1000 K and progressively extracting Lithium to mimic battery charging. We observe a clear shift over time: at first, the manganese (Mn) atoms supply the electrons needed for charging, but as more lithium is removed, oxygen atoms are forced to give up electrons instead (cationic to anionic redox), as shown by the shift to less negative Bader charges over time (Fig. 3(d)). This destabilises the structure with oxygen atoms pairing up to form O2 dimers (Fig. 3(d), inset). Notably, this comprehensive picture of the cationic-to-anionic redox transition and lattice degradation naturally emerges from the multi-task predictions, without any task-specific training on battery materials.
Next steps
With experimental validation, substantial performance improvements, and new multi-task capabilities, MatterSim is advancing toward more practical, decision-relevant use in materials design. Together, these developments are helping materials scientists move more quickly from large-scale computational screening to targeted experimental follow-up and decision-relevant scientific workflows. We are excited to see how the materials science community applies these advances in their own domains.
We look forward to continued collaboration as MatterSim is tested, extended, and integrated into real-world materials discovery pipelines.
MatterSim updated
MatterSim-MT Pre-Print
Acknowledgements
This work is the product of a highly collaborative and interdisciplinary effort led by Microsoft Research AI for Science in partnership with Microsoft Research Accelerator and collaborators at the University of Texas Dallas (supported by MSR Accelerator), University of Illinois Urbana-Champaign and University of California Davis. Contributors to this work include Han Yang, Xixian Liu, Chenxi Hu, Yichi Zhou, Yu Shi, Chang Liu, Junfu Tan, Jielan Li, Guanzhi Li, Qian Wang, Yu Zhu, Zekun Chen, Shuizhou Chen, Fabian Thiemann, Claudio Zeni, Matthew Horton, Robert Pinsler, Andrew Fowler, Daniel Zügner, Tian Xie, Lixin Sun, Yicheng Chen, Lingyu Kong, Yeqi Bai, Deniz Gunceler, Frank Noé, Hongxia Hao, Ziheng Lu, Zixin Zhai, Mengfan Wu, Haoke Qiu, Mingfa Tang, Tie-Yan Liu, Haiguang Liu, Tao Qin, David G. Cahill, Bing Lv, Davide Donadio, Shoko Ueda, and Kenji Takeda.
Opens in a new tabThe post Advancing AI for materials with MatterSim: experimental synthesis, faster simulation, and multi-task models appeared first on Microsoft Research.
関連記事
今日のまとめ
AI日報で今日の重要ニュースをまとめ読み